Introduction
Long QT syndrome (LQTS) and Brugada syndrome (BrS) are well-known congenital channelopathies that can cause sudden cardiac death (SCD). Although various genetic mutations are associated with LQTS and BrS, an electrocardiogram (ECG) is typically preferred to genetic analysis as a diagnostic tool in clinical practice. However, care must be taken when using ECG in the diagnosis of LQTS or BrS, because the characteristic ECG changes of these syndromes are not observed consistently. The ECG pattern can be altered by various factors such as an aborted SCD, the autonomic nervous system, medication, circadian rhythms, and electrolyte imbalance.
Here, we report our experience with a family in which the proband displayed an ECG pattern that initially suggested the presence of LQTS but was ultimately diagnosed as BrS, based on the findings of serial ECG follow-up, a drug provocation test, and genetic analysis.
Case
Proband
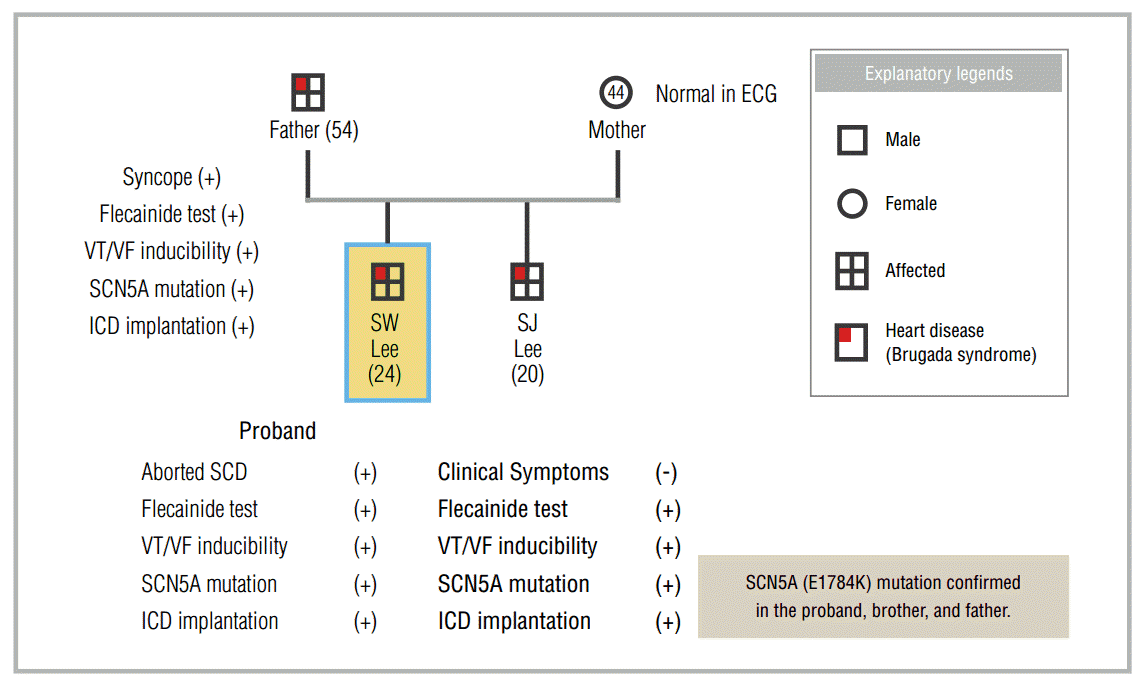
A 24-year-old Air Force sergeant was successfully resuscitated after SCD and was transferred to the Armed Forces Capital Hospital (AFCH). He had collapsed when performing his regular activities and received immediate cardiopulmonary resuscitation (CPR) and automated external defibrillation, which were provided by his colleagues. The ECG recorded by the automated external defibrillator showed ventricular fibrillation. After resuscitation, the proband developed pulmonary edema, for which he was treated at another hospital. The structure and function of the heart were found to be normal, and no abnormalities were revealed with other tests (treadmill ECG test and Holter ECG). Two weeks after the aborted SCD, a ventricular tachycardia (VT) study was performed. In the VT study, fast polymorphic VT (cycle length, 90-180 ms) was reproducibly induced during the double ventricular extrastimulation test (DVEST) at 500/260/220 ms.
The first ECG performed at the AFCH indicated a heart rate of 60 beats/min in normal sinus rhythm, a corrected QT interval (QTc) of 520 ms, and narrow-based and deeply inverted symmetric T waves in leads II, III, aVF, and V3-6 (Figure 1A). LQT3 was suspected, even though symmetric T wave inversion is unusual in this condition, because of the presence of QTc prolongation, narrow-based T waves, and history of aborted SCD. An implantable cardioverter defibrillator (ICD) was implanted for secondary prevention of SCD, and VT was reproducibly induced with DVEST in a shock test performed after ICD implantation.
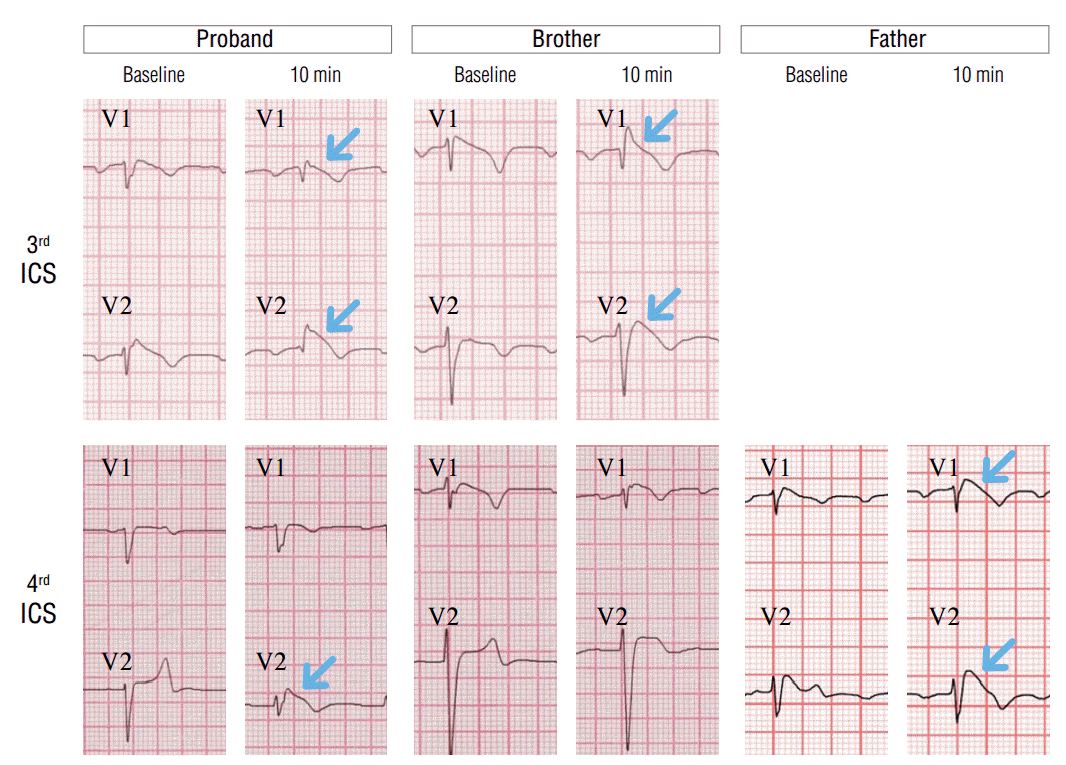
Two weeks after ICD implantation, T wave inversion and QTc prolongation had disappeared, and ST changes with type 1 Brugada pattern was observed in V1, 2 leads of the follow-up ECG taken from the 3rd intercostal space (Figure 1B). In addition to these changes, the flecainide test was performed, and ST changes from type 3 to type 1 Brugada pattern were observed, which confirmed the diagnosis of BrS in the proband (Figure 2).
Family Screening
ECGs were performed in all family members. The ECG of the mother indicated a heart rate of 63 beats/min in normal sinus rhythm and a normal QTc of 414 ms. The ECG of the father indicated a heart rate of 60 beats/min with normal sinus rhythm, a PR interval of 220 ms, QTc of 453 ms, and lack of inversion of the T wave. ST elevation with a coved pattern was observed in V1, and ST elevation with a saddle back pattern was observed in V2, which is suggestive of BrS. Because he had experienced a passing spell requiring brief CPR during his post-office work approximately 1 year ago, a provocation test was performed with intravenous flecainide. After intravenous flecainide infusion, the ECG changes became more prominent (Figure 2).
The ECG of the younger brother demonstrated marked sinus bradycardia with sinus arrhythmia (42 beats/min), an increased PR interval of 220 ms, and a prolonged QTc of 460 ms. Although the T wave was not inverted, it had a tall and narrow-based morphology, which may suggest presence of LQTS. However, BrS may also be suspected based on the J point elevation with a coved pattern in V1 and ST elevation with a saddle back pattern in V2.
Although the VT induction test is of limited value in the diagnosis of BrS, it was performed in the father and brother. As observed in the VT induction test of the proband, fast polymorphic sustained VT was reliably induced. The pacing protocols were single ventricular extrastimulation at 400/260 ms in the father and DVEST at 600/300/210 ms in the brother.
It was subsequently confirmed that the same SCN5A gene mutation (E1784K) was present in all 3 patients. Finally, ICDs were implanted in all 3 patients who were then followed (Figure 3).
To summarize, attempting to determine the cause of the aborted SCD led to the diagnosis of BrS in the asymptomatic brother and the father who had experienced syncope as reported during a family screening ; they were treated with ICD implantation. This case emphasizes the importance of a careful serial ECG follow-up and genetic analysis during family screening of an SCD proband with atypical clinical features.
Discussion
Congenital LQTS and BrS are characterized by ECG abnormalities that are accompanied by SCD and were first described a few decades ago. Congenital LQTS is characterized by the prolongation of ventricular repolarization that predisposes the patient to torsades de pointes and SCD. BrS is characterized by syncope and SCD from ventricular tachyarrhythmias and right precordial ST segment elevation in the ECG. Although both syndromes were described as ECG predictors of SCD before the era of molecular and genetic diagnosis, it became apparent that both disease entities result from abnormalities in ion channels and proteins that alter the ventricular repolarization phase. To date, more than 100 SCN5A mutations have been associated with BrS or LQTS3 [1-5]. Although all the mutations associated with BrS or LQTS have not been fully characterized, large bodies of genetic and molecular evidence have improved our understanding of these syndromes.
The E1784K mutation, originally reported by Wei, et al., is the most prevalent LQTS3 mutation, and is present in approximately 15.4-34% of LQTS3 probands [6-8]. Priori, et al. reported that phenotypical overlap with BrS is relatively common, and Na channel blocker therapy that is administered for LQTS3 can unmask or exacerbate the ST elevation of BrS [9]. According to the biophysical assessment of this mutation, a negative shift in inactivation and an enhanced tonic block appear to be common properties of this BrS/LQTS3 overlapping phenotype. If ECG manifestations are inconclusive, careful serial ECG follow-up and genetic analysis can provide additional information, as in the present case, which can improve clinical decision-making regarding therapeutic modalities and family screening.
In conclusion, careful serial ECG follow-up and genetic or molecular assessment, in addition to their diagnostic and screening value, can improve the mechanistic understanding of congenital channelopathies such as LQTS and BrS, especially when the clinical and ECG presentations are atypical. This case demonstrated the importance of careful serial ECG follow-up and genetic analysis during family screening of the SCD proband with atypical clinical features.