|
|
International Journal of Arrhythmia 2014;15(2): 4-16.
|
 |
| ORIGINAL ARTICLES |
Arrhythmogenic Mechanisms of
Autoimmune Myocarditis Associated with
Inflammation and Ca2+/Calmodulin-
Dependent Protein Kinase II Activation |

|
|
|
|

Introduction
Myocarditis and subsequent dilated cardiomyopathy
(DCM) are major causes of heart failure in young patients.1 Myocarditis is characterized by infiltration
of inflammatory cells into the myocardium,
with consequent loss of myocytes and development of
fibrosis and necrosis.2 In a significant fraction of patients,
the loss of cardiomyocytes leads to ventricular
remodeling, permanent ventricular wall dysfunction,
DCM, heart failure, and/or arrhythmias. Myocarditis
is induced by a variety of means, including genetic
susceptibility, toxins, viruses, bacteria, and parasites.
3-4 It is associated with arrhythmias and sudden
death, and the treatment of ventricular arrhythmias
is critical for improvement in patient prognosis.5
Experimental autoimmune myocarditis (EAM) in
the rat is a unique and useful model for understanding
giant cell myocarditis and subsequent DCM.6 EAM
rats are highly susceptible to ventricular arrhythmia
and prolonged action potential duration (APD). Reduced
expression of Ito-related molecules, including
Kv4.2, Kv1.5, frequenin and KChIP2 is considered
to play a key role in ventricular remodeling, and to
cause the characteristic clinical findings of EAM.7-8
Cardiac inflammation - a hallmark of myocarditis - is known to increase oxidative stress. Niwano et al.9 demonstrated previously that the anti-oxidant
N-acetylcysteine suppresses ventricular remodeling
in EAM rats, suggesting that oxidative stress plays a
role in remodeling and the development of myocarditis
itself during the acute phase of myocarditis. It was
recently reported that oxidative stress can activate
Ca2+/calmodulin-dependent protein kinase II (CaMKII),10 prolong APD and induce afterdepolarization in
cardiomyocytes.11 Therefore, we hypothesized that
EAM can induce arrhythmia via CaMKII activation
caused by inflammation and oxidative stress. To test
this hypothesis, we evaluated arrhythmogenic events
and survival in an EAM model. Arrhythmogenic
mechanisms underlying EAM were further investigated
in Langendorff-perfused isolated hearts, and the level of inflammation and CaMKII activation in
myocarditis were subsequently assessed. Finally, we
confirmed that arrhythmogenic events and CaMKII
activation were suppressed following pretreatment
with an anti-inflammatory corticosteroid agent.
Materials and methods
This study protocol was approved by the Institutional
Animal Care and Use Committee of Yonsei
University College of Medicine and Cardiovascular
Research Institute, and conforms to the guidelines of
the American Heart Association.
Induction of experimental autoimmune myocarditis
Purified cardiac myosin (M0531, Sigma Aldrich,
Schnelldorf, Germany)12 was emulsified in an equal
volume of complete Freund’s adjuvant (BD biosciences,
Heidelberg, Germany) supplemented with
mycobacterium tuberculosis H37 Ra (Difco, Detroit,
USA) at a concentration of 10 mg/mL. Six-weekold
male Lewis rats were immunized by subcutaneous
injection of 2 mg purified cardiac myosin in each rear
footpad on days 1 and 8 (Myo group; n=15). Control
rats received injections of 0.5 mL complete Freund’s
adjuvant in the same manner (Control group; n=15).
In a separate group of 15 rats, 6 mg steroid was administered
simultaneously with cardiac myosin on
days 1 and 8 (MyoS group; n=15). Ambulatory Holter
monitoring was performed using a telemetric system
(Telemetry Research, Auckland, New Zealand).
Histology and inflammatory cytokine assay
Following measurement of hemodynamic parameters,
hearts were immediately excised and weighed;heart weight to body weight ratios were calculated.
Hearts were stained with hematoxylin and eosin, and
Masson’s trichrome stain. Immunostaining was performed
using a tumor necrosis factor-α (TNF-α) antibody
to evaluate the degree of inflammation.
Blood was obtained from the abdominal aorta of
each rat on day 21. An enzyme-linked immunosorbent
assay (ELISA) was performed to determine serum
levels of high-mobility group box protein 1 (HMGB1),
interleukin 6 (IL-6), and TNF-α. Serum protein levels
were quantified with kits for HMGB1 (IBL International,
Hamburg, Germany), IL-6 (R&D System,
Minneapolis, MN, USA) and TNF-α (R&D System,
Minneapolis, MN, USA), according to manufacturer's instructions.
Optical mapping
On the 21st day after the initial immunization, rats
(250-300 g) were anesthetized by intraperitoneal injection
of ketamine (80 mg/kg) and xylazine (4 mg/
kg). Chests were opened via median sternotomy and
the hearts were rapidly excised and immersed in cold
Tyrode’s solution (composition in mmol/L: 125 NaCl,
4.5 KCl, 0.25 MgCl2, 24 NaHCO3, 1.8 NaH2PO4, 1.8
CaCl2, and 5.5 glucose). The ascending aorta was immediately
cannulated and perfused with Tyrode’s solution
prewarmed to 37°C and equilibrated with 95%
O2 and 5% CO2 to maintain a pH of 7.4. Coronary
perfusion pressure was maintained between 80 and
95 mmHg. Two widely spaced bipolar electrodes were
used for continuous pseudo-ECG monitoring.
For optical recording, cardiac contractility was inhibited
with 10-17 μmol/L blebbistatin.13 Hearts were
stained with di-4-ANEPPS (Invitrogen, California,
USA) and excited with quasi-monochromatic light
(520 ± 30 nm) from two green LED lamps. Emitted
light was collected by an image-intensified chargecoupled device camera (Dalsa Inc., Waterloo, Canada)
with a 610-nm long pass filter. Data were gathered
at 3.75-ms sampling intervals, acquiring simultaneously
from 125 × 125 pixels, each 0.08 mm × 0.08
mm. The mapped area included parts of the right and
left ventricular free walls.
Optical recordings were performed during steadystate
and programmed stimulation. Programmed
stimulation was induced with bipolar electrodes
at the lateral side of the left ventricle (LV). Pacing
was initiated at a cycle length (CL) of 300 ms, using
stimuli of twice the pacing threshold, and was
subsequently reduced in decrements of 10 ms until
2:1 capture was achieved. APD at 90% repolarization
(APD90) was measured at the base and apex of the
LV. APD dispersion was defined as the difference between
maximum and minimum APD. After the initial
electrophysiological study, we attempted to induce
ventricular tachycardia (VT) or ventricular fibrillation
(VF) using a standard pacing technique (burst pacing
at CLs down to 70 ms). All sustained (>30 s) and
non-sustained VT or VF episodes were documented.
Optical mapping and VT induction studies were performed
in 6 rats from each group.
Immunoblot analysis of Ca2+ handling proteins
Immunoblotting for CaMKII, ryanodine receptor
type 2 (RyR2), phospholamban (PLB) and the phosphorylated
form of each protein was performed using
the following monoclonal antibodies: anti-CaMKII
and anti-p-CaMKII (1:1,000; Santa Cruz Biotechnology);
anti-RyR2 (1:1000; Abcam Reagents); antip-
RyR2 (1:1000; Badrilla); and anti-p-PLB (1:1000;
Santa Cruz Biotechnology). Targeted antigens were
visualized by labeling with corresponding HRP-conjugated
secondary IgG (1:5,000; Santa Cruz Biotech nology) followed by enhanced chemiluminescence
assay (ECL Plus, Amersham, Piscataway, NJ). Blots
were scanned, and band intensity was quantified using
the Image J software.
Statistical analysis
Data are expressed as mean ± SEM. The Student’s
t-test with Bonferroni correction was used to compare
the means of two numeric values. The Pearson's
chi-square test was used to compare two categorical variables. Paired t-tests were used to compare the
means of maximum slope of restitution curves between
baseline and myocarditis. p values <0.05 were
considered statistically significant.

Results
Arrhythmia and survival of EAM
For the control, Myo and MyoS groups, heart
weights were 1.2 ± 0.1 g, 1.7 ± 0.1 g, and 1.3 ± 0.1
g, respectively (Figure 1A). Heart weight was significantly greater in the Myo group than in the control
(p=0.002) and MyoS (p=0.03) groups. Compared with
controls, the ratio of heart weight to body weight was
significantly increased in the Myo group (5.2 ± 0.5
vs. 7.1 ± 0.4, p<0.001), but not in the MyoS group
(5.8 ± 0.3, p=0.31). Figure 1B shows histological
findings 21 days after the initial immunization with
cardiac myosin. While no inflammatory changes were
observed in the control rats, there was infiltration of
inflammatory cells in Myo rats, including giant cells
mainly in the right ventricle and epicardial layer of
LV. Severe interstitial fibrosis and increased TNF-α
expression were also observed in Myo rats. Infiltration of inflammatory cells, fibrosis and increased TNF-α
expression were attenuated in the MyoS group. Figure
1C shows the Kaplan-Meier survival curves for
the three groups. While 5 out of 15 rats (33%) in the
Myo group died suddenly at 17 ± 2 days after induction
of myocarditis, no rats and one rat (6%) died in
the control and MyoS groups, respectively. The Myo
group had a lower cumulative survival rate than the
control group (p=0.02). Figure 1D shows the various
arrhythmias recorded by ambulatory Holter monitoring
in the Myo group. VT, sinus pause and atrioventricular
block were observed in 4 rats in the Myo
group, 3 in the control group, and 3 in the MyoS group. While arrhythmias were not observed in control
rats, they were observed in 5 of the 9 surviving
rats (56%) in the Myo group, and in none of the 15
rats in the MyoS group (p=0.03).
Inflammatory markers in EAM
Serum levels of HMGB1, IL-6 and TNF-α are presented
in Supplementary Table 1. Compared with
controls, serum levels of HMGB1, IL-6 and TNF-α
in the Myo group were increased 1.1-fold (p<0.001),
2.1-fold (p<0.001), and 4.0-fold (p<0.001), respectively. Conversely, no significant change in expression
levels was observed in the MyoS group.
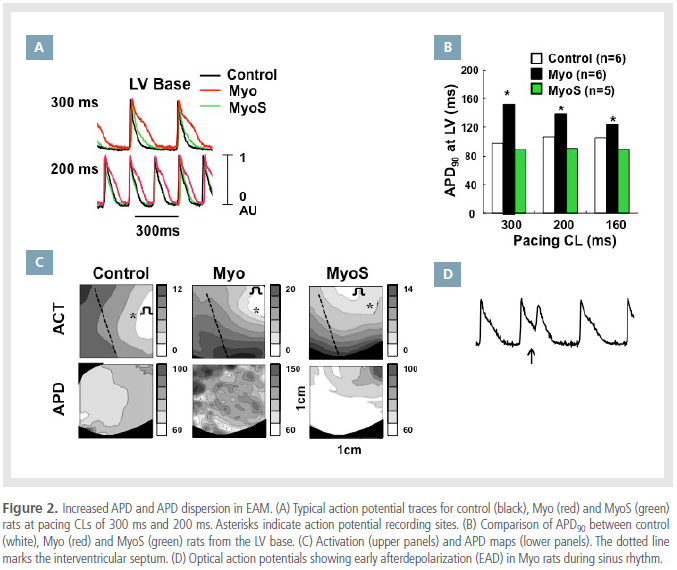
Increased APD and APD dispersion in EAM
Optical mapping was performed in 6 rats from each
group. Figure 2A shows the action potential traces
obtained from the base of the LV during pacing CLs
of 300 ms and 200 ms in Langendorff-perfused rat
hearts. Mean APD in the Myo group was relatively
prolonged compared to the control and MyoS groups.
A comparison of mean APD90 measured at LV in all three groups is presented in Figure 2B. Mean APD90
at a pacing CL of 300 ms was significantly longer
in the Myo than in the control group (152 ± 52 ms
vs. 98 ± 7 ms, p=0.03). APD90 was not prolonged in
the MyoS group (89 ± 7 ms, p=1.0), however. Mean
APD90 was also longer in Myo rats than control and
MyoS rats at pacing CLs of 200 and 160 ms. Figure
2C shows corresponding activation and repolarization
maps. The conduction time of both ventricles was
prolonged in the Myo group compared with control
and MyoS groups. APD dispersion was similarly increased
in the Myo group (23.6 ± 3.6 ms) compared
to controls (5.8 ± 2.0 ms, p<0.001), though this
increase was attenuated in the MyoS group (10.5 ± 1.7 ms, p<0.001). Figure 2D shows the optical action
potentials of early afterdepolarization (EAD) in Myo
rats during sinus rhythm.
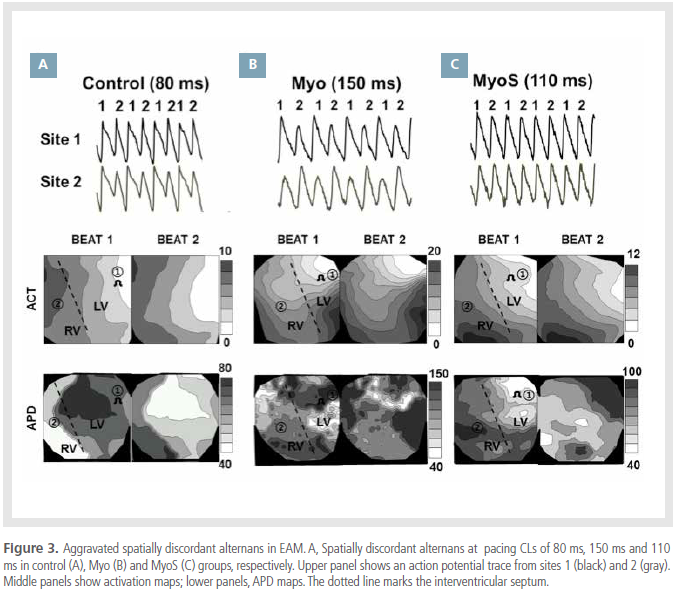
APD alternans and ventricular arrhythmia in EAM
APD alternans were evaluated in all 3 groups (Figure
3). Discordant alternans were observed during a
pacing CL of 80 ms in control rats (Figure 3A), 150 ms
in Myo rats (Figure 3B) and 110 ms in MyoS rats (Figure
3C). Compared with controls, the pacing CL required
to induce discordant alternans in Myo rats was
increased from 80 ± 9 ms to 178 ± 22 ms (p<0.001);no significant difference was observed between control
and MyoS rats (114 ± 5 ms, p=0.06). These findings
suggest that spatially discordant alternans were
more easily induced in the Myo group, than in other
groups. Conduction block was also more frequently
observed at longer pacing CLs in the Myo group
(107 ± 10 ms) than in the control group (73 ± 6 ms,
p<0.001), which was not significantly different from
the MyoS group (84 ± 10 ms, p=0.24).
Ventricular arrhythmias were evaluated in 6 rats
from each group. For the control, Myo and MyoS
groups, triggered activity was observed in 1 (17%), 5
(83%) and 2 (33%) rats, respectively. Of the 6 rats in each group, ventricular arrhythmias were induced in
0, 6 (100%) and 1 (27%), respectively. The Myo group
exhibited triggered activity (p=0.02) and ventricular
arrhythmia (p=0.03) more frequently than the control
group. Conversely, the MyoS group showed no
significant difference in triggered activity (p=0.55) or
ventricular arrhythmia (p=0.57) compared with controls.

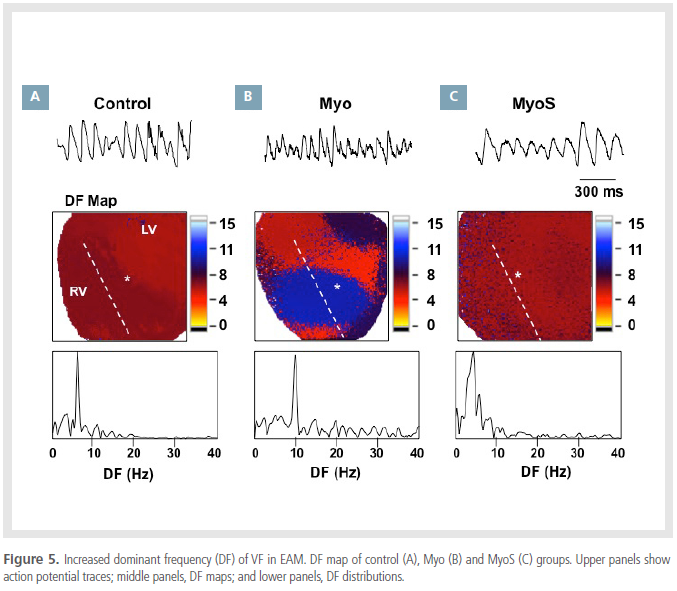
Increased APD restitution slope and dominant frequency in EAM
Figure 4 shows a typical example of an APD restitution curve measured at the LV base and apex. For
the control, Myo and MyoS groups, the maximum
APD restitution slopes were 0.23 ± 0.09, 0.70 ± 0.10
and 0.28 ± 0.04 at the LV base, respectively. Slopes
at the LV apex were 0.26 ± 0.07, 1.19 ± 0.11 and
0.24 ± 0.09, respectively. The APD restitution slopes
in the Myo group were steeper than controls at both
the LV base (p<0.001) and apex (p<0.001). A significant
increase relative to control was not observed
in the MyoS group, however (p=1.00).
The dominant frequency (DF) maps of VF episodes
were evaluated in the 3 groups. DF was 6 Hz in the
control group (Figure 5A), 8 Hz in the Myo group(Figure 5B) and 5 Hz in the MyoS group (Figure 5C).
Compared with the control group, mean DF was significantly
higher in the Myo group (8.1 ± 1.0 vs. 6.3
± 0.3 Hz, p=0.006) but not in the MyoS group (5.8
± 0.5 Hz, p=0.82).
Increased p-CaMKII, ryanodine and pphospholamban in EAM
Figure 6 shows a Western blot of Ca2+ handling
proteins. Compared with controls, p-CaMKII,
RyR2, p-RyR2 and p-PLB were increased 2.5-fold
(p<0.001), 2.9-fold (p<0.001), 5.1-fold (p<0.001) and 2.3-fold (p<0.001), respectively, in the Myo
group. This significant increase was not observed in
the MyoS group. CaMKII expression was not different
between the 3 groups.
Discussion
The salient findings of this study were four-fold.
Firstly, EAM rats were found to exhibit fatal arrhythmia
and decreased survival relative to controls.
Secondly, the EAM model was characterized by prolonged
APD with increased APD dispersion, easily
inducible spatially discordant alternans, steeper APD
restitution slopes and increased ventricular arrhythmia.
Thirdly, increased activity of calcium handling
proteins, including p-CaMKII, p-RyR and p-PLB, is
induced in the EAM model. Finally, EAM-related arrhythmia
and the activation of calcium handling proteins
were attenuated by pretreatment with an antiinflammatory
steroid. Our results indicate that EAM
can induce arrhythmia via CaMKII activation under
conditions of inflammation and oxidative stress.
Ventricular arrhythmias and decreased survival in EAM rats
Ventricular arrhythmia is a significant cause of
death, along with heart failure, in acute myocarditis.
The myosin-induced EAM rat is one of the animal
models used to study the events that occur in human
giant cell myocarditis.6,14-17 The EAM model comprises
an acute inflammatory phase evoked 2 weeks after
myosin injection, and a subsequent recovery phase
initiated around the 25th day after injection, followed
by a dilated cardiomyopathy-like phase associated
with chronic heart failure. In this study, 9 out of 15
EAM rats (60%) exhibited cardiac events, including
sudden death and arrhythmia. These results suggest that the low survival rate associated with the EAM
model is likely related to the development of arrhythmia.
Interestingly, in addition to VT, sinus dysfunction
and atrioventricular block were also commonly
observed in the EAM model.
Increased repolarization gradient and CaMKII activation in EAM
APD was prolonged in myosin-induced EAM.7 This
prolongation may be explained by an initial reduction
in Ito-related currents, following downregulation
of Kv4.2, Kv1.5, frequenin and KChIP2.7-8 The
role of Ca2+ handling proteins in EAM has not yet
been established, however. Our results indicate that
the activation of Ca2+ handling proteins may play an
important role in EAM-induced arrhythmia. We observed
increased expression of CaMKII and phosphorylated
CaMKII following induction of myocarditis,
suggesting that CaMKII activation may facilitate APD
prolongation, representing a novel arrhythmogenic
mechanism of EAM.
EAM-associated inflammation increases oxidative
stress. In addition to activation by elevated intracellular
Ca2+ levels (following β-adrenergic receptor
stimulation18) CaMKII activity is also known to be
enhanced under pro-oxidant conditions.10,19-20 Oxidation
of paired regulatory domain methionine residues
sustains CaMKII activity in the absence of Ca2+/
CaM.10 H2O2-induced afterdepolarizations depend on
both impaired INa inactivation, to reduce repolarization
reserve, and enhanced ICa,L, to reverse repolarization,
both of which are facilitated by CaMKII activation.11
Consistent with elevated p-CaMKII, p-RyR2 and
p-PLB levels were also increased in EAM. The RyR,
or calcium release channel, on the sarcoplasmic reticulum
is the major source of calcium required for
excitation-contraction coupling in cardiac muscle. Hyperphosphorylation of RyR2 results in defective
function due to increased sensitivity to Ca2+-induced
activation.21
Until now, it has not been clear whether APD prolongation
in myocardial cells is homogeneous or heterogeneous
in nature, because previous studies have
only evaluated a single site in the heart using a macroscopic
electrophysiological technique. In this study,
using optical mapping, we observed heterogeneous
APD prolongation and increased APD dispersion in
EAM. Although APD prolongation is the main mechanism
of long QT syndrome, enhanced dispersion of
repolarization is critical for the induction of fatal arrhythmia.22 Transmural and apicobasal dispersion of
repolarization was shown to be responsible for the
initiation of reentrant activation in long QT syndrome
patients. It was reported that numerous mammalian
species, including humans, exhibit apex-base differences
in cardiac repolarization.23 Increased spatial
dispersion of repolarization across the anterior epicardial
surface was also demonstrated to represent
the electrical basis for spontaneous malignant arrhythmias
in long QT type 2 rabbits.24
Mechanisms of ventricular arrhythmia in EAM
Spatially discordant alternans were easily induced
in EAM. These cause an increase in the spatial dispersion
of repolarization, and are thought to result in
T-wave alternans.25 T-wave alternans are precursors
of cardiac electrical instability, and consequently
sudden cardiac death.26 Spatially discordant alternans
can be explained by the increased steepness of APD
restitution slopes in EAM. A steep slope of electrical
restitution facilitates the breakup of single spiral
waves into multiple spiral waves, a process that may
account for the transition from VT to VF.27-28 A slope of electrical restitution ≥1 is especially associated
with VF.29 Furthermore, the increased prevalence of
discordant alternans may also have been the result
of fibrosis and reduced gap junction conductance in
EAM; these have previously been shown to lower the
threshold for spatially discordant alternans.30-31 Finally,
the development of discordant alternans may
also be related to altered expression and activity of
Ca2+ handling proteins. The net effects of EAM remodeling
promote Ca2+ alternans via phosphorylation
of RyRs and CaMKII signaling to increase their Ca2+
sensitivity (increasing both gain and leak).32-33
Attenuation of EAM-related arrhythmia by anti-inflammatory therapy
In this model, the overexpression of inflammatory
cytokines, such as HMGB-1, TNF-α and IL-6
can induce myocardial damage and possibly cause
ventricular remodeling.6,34 Because these inflammatory
cytokines are strong inducers of nitric oxide and
reactive oxygen species, they may promote cardiac
injury and electrical remodeling through precipitation
of hyper-oxidative conditions. Niwano et al.9
previously reported that the anti-oxidant N-acetylcysteine
suppressed ventricular remodeling in EAM
rats. Concordantly, we have demonstrated that antiinflammatory
steroid therapy suppresses infiltration
of inflammatory cells and EAM-induced electrical
remodeling. Moreover, steroid pretreatment was associated
with improved survival. This indicates the
importance of inflammation and oxidative stress in
remodeling and the progression of myocarditis.
Study limitations
In this study, we induced myocarditis by injection
of cardiac myosin. Therefore, the arrhythmogenic mechanisms delineated may not be representative of
those underlying myocarditis caused by viral infection
or other etiologies. Nonetheless, autoimmunization to
myosin may represent a final common pathway of
myocarditis. Moreover, inflammation and oxidative
stress are frequently observed in diseased hearts.
Secondly, electrophysiological tests and optical
mapping were performed on the 14th day after initial
immunization. To identify individual differences
after induction of myocarditis, it would be useful to
perform optical mapping at different time points after
immunization. Because an electrophysiological
test could not be performed in rats that succumbed
to sudden death, we could not provide direct evidence
that APD dispersion and discordant alternans were
related to mortality.
Finally, ventricular fibrillation was recorded during
sudden death in rats with myocarditis. However, because
Holter monitoring was performed in only 5 rats
that died suddenly, we cannot rule out the possibility
that the AV block was responsible for sudden death.
Conclusion
EAM-induced arrhythmia is associated with increased
repolarization dispersion and spatially discordant
alternans. These electrical changes may be
related to altered Ca2+ handling protein activity, including
phosphorylation of CaMKII and RyR2. These
arrhythmogenic effects are associated with increased
inflammation and oxidative stress, and in this study
were attenuated by anti-inflammatory therapy.
Sources of funding
This study was supported in part by research grants
from Yonsei University College of Medicine (8-2011-
0250, 7-2011-0758, 7-2011-0702, 7-2011-0015), a grant from the Korean Heart Rhythm Society (2011-
3) and the Basic Science Research Program (2012-
0007604, 2012-045367) through the National Research
Foundation of Korea, funded by the Ministry
of Education, Science and Technology.
Disclosure
None.
References
- Eriksson U, Penninger JM. Autoimmune heart failure: New understandings of pathogenesis.
Int J Biochem Cell Biol.
2005;37:27-32.
- Feldman AM, McNamara D. Myocarditis. N Engl J Med.
2000;343:1388-1398.
- Kawai C. From myocarditis to cardiomyopathy: Mechanisms of inflammation and cell death: Learning from the past for the future.
Circulation.
1999;99:1091-1100.
- Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy.
Adv Immunol.
2008;99:95-114.
- Levi D, Alejos J. Diagnosis and treatment of pediatric viral myocarditis.
Curr Opin Cardiol.
2001;16:77-83.
- Kodama M, Matsumoto Y, Fujiwara M, Masani F, Izumi T, Shibata A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction.
Clin Immunol Immunopathol.
1990;57:250-262..
- Saito J, Niwano S, Niwano H, Inomata T, Yumoto Y, Ikeda K, Inuo K, Kojima J, Horie M, Izumi T. Electrical remodeling of the ventricular myocardium in myocarditis: Studies of rat experimental autoimmune myocarditis.
Circ J.
2002;66:97-103.
- Wakisaka Y, Niwano S, Niwano H, Saito J, Yoshida T, Hirasawa S, Kawada H, Izumi T. Structural and electrical ventricular remodeling in rat acute myocarditis and subsequent heart failure.
Cardiovasc Res.
2004;63:689-699.
- Niwano S, Niwano H, Sasaki S, Fukaya H, Yuge M, Imaki R, Machida Y, Izumi T. N-acetylcysteine suppresses the progression of ventricular remodeling in acute myocarditis: Studies in an experimental autoimmune myocarditis (EAM) model.
Circ J.
2011;75:662-671..
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of camkii by methionine oxidation.
Cell.
2008;133:462-474.
- Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stressinduced afterdepolarizations and calmodulin kinase ii signaling.
Circ Res.
2009;104:79-86.
- Inomata T, Hanawa H, Miyanishi T, Yajima E, Nakayama S, Maita T, Kodama M, Izumi T, Shibata A, Abo T. Localization of porcine cardiac myosin epitopes that induce experimental autoimmune myocarditis.
Circ Res.
1995;76:726-733.
- Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW, Efimov IR. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts.
Heart Rhythm.
2007;4:619-626.
- McFalls EO, Hosenpud JD, McAnulty JH, Kron J, Niles NR. Granulomatous myocarditis. Diagnosis by endomyocardial biopsy and response to corticosteroids in two patients. Chest. 1986;89:509-511.
- Davidoff R, Palacios I, Southern J, Fallon JT, Newell J, Dec GW. Giant cell versus lymphocytic myocarditis. A comparison of their clinical features and long-term outcomes. Circulation. 1991;83:953-961.
- Kodama M, Hanawa H, Saeki M, Hosono H, Inomata T, Suzuki K, Shibata A. Rat dilated cardiomyopathy after autoimmune giant cell myocarditis. Circ Res. 1994;75:278-284.
- Izumi T, Takehana H, Matsuda C, Yokoyama H, Kohno K, Suzuki K, Inomata T. Experimental autoimmune myocarditis and its pathomechanism. Herz. 2000;25:274-278.
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr., Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase ii inhibition protects against structural heart disease. Nat Med. 2005;11:409-417.
- Howe CJ, Lahair MM, McCubrey JA, Franklin RA. Redox regulation of the calcium/calmodulin-dependent protein kinases. J Biol Chem. 2004;279:44573-44581.
- Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of camkiideltac is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. 2007;282:10833-10839.
- Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365-376.
- Antzelevitch C. Ionic, molecular, and cellular bases of qt-interval prolongation and torsade de pointes. Europace. 2007;9 Suppl 4:iv4-15.
- Szentadrassy N, Banyasz T, Biro T, Szabo G, Toth BI, Magyar J, Lazar J, Varro A, Kovacs L, Nanasi PP. Apico-basal inhomogeneity in distribution of ion channels in canine and human ventricular myocardium. Cardiovasc Res. 2005;65:851-860.
- Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M, Koren G. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long qt syndrome. J Clin Invest. 2008;118:2246-2259.
- Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking t-wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385-1394.
- Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. Engl J Med. 1994;330:235-241.
- Karma A. Electrical alternans and spiral wave breakup in cardiac tissue. Chaos. 1994;4:461-472.
- Witkowski FX, Leon LJ, Penkoske PA, Giles WR, Spano ML, Ditto WL, Winfree AT. Spatiotemporal evolution of ventricular fibrillation. Nature. 1998;392:78-82.
- Riccio ML, Koller ML, Gilmour RF, Jr. Electrical restitution and spatiotemporal organization during ventricular fibrillation. Circ Res. 1999;84:955-963.
- Pastore JM, Laurita KR, Rosenbaum DS. Importance of spatiotemporal heterogeneity of cellular restitution in mechanism of arrhythmogenic discordant alternans. Heart Rhythm. 2006;3:711-719.
- Pastore JM, Rosenbaum DS. Role of structural barriers in the mechanism of alternans-induced reentry. Circ Res. 2000;87:1157-1163.
- Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69-98.
- Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Betaadrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulindependent protein kinase. Circ Res. 2007;100:391-398.
- Okura Y, Yamamoto T, Goto S, Inomata T, Hirono S, Hanawa H, Feng L, Wilson CB, Kihara I, Izumi T, Shibata A, Aizawa Y, Seki S, Abo T. Characterization of cytokine and inos mrna expression in situ during the course of experimental autoimmune myocarditis in rats. J Mol Cell Cardiol. 1997;29:491-502.
|
|
|








