Introduction
Infarct healing is a complex biological process that occurs during the first 2 weeks after myocardial infarction (MI) [1-3]. The quality of healing influences the outcome; impaired healing leads to infarct expansion and accelerated left ventricular remodeling [4,5]. Reperfusion strategies, specifically thrombolytic and antiplatelet therapies, have remarkably improved the clinical outcome in patients with acute MI. However, beneficial effects of reperfusion therapies are limited by the amount of ischemic damage that occurs before reperfusion. Several studies have focused on developing adjunctive pharmacological interventions to enhance the myocardial tolerance to ischemia/reperfusion (IR) injury.
The receptor for advanced glycation end products (RAGE), a multiligand receptor of the immunoglobulin superfamily, interacts with distinct families of ligands that mediate diverse functions in a broad array of cell types, including cellular migration and activation of proinflammatory mechanisms [6]. Initially, RAGE was described as a receptor for advanced-glycation end products (AGEs), which accumulate under hyperglycemic conditions [6]. Subsequent studies have shown that S100/calgranulins, amphoterin, and amyloid peptide can also bind to RAGE, causing the activation of signal transduction and modulation of gene expression [7-9]. In chronic disease states such as diabetes, immune/inflammatory foci, and neurodegenerative disorders, upregulation of RAGE was associated with tissue injury [8,9]. Recently, RAGE was shown to modulate (1) the response to acute nerve injury [10] and (2) cell death-promoting mechanisms in severe hepatectomy [11]. Furthermore, the RAGE ligand S100B was shown to modulate cardiac hypertrophy and remodeling after MI [12].
RAGE silencing was reported to decrease MI [13]. However, the role of the RAGE in the treatment of arrhythmia is unknown. In this study, we evaluated whether RAGE decreases IR injury by using a rat MI model. Second, we evaluated whether RAGE could suppress arrhythmia in the rat IR model.
Methods
Animal Studies
All studies involving rats were approved by the Institutional Animal Care and Use Committee of Yonsei University College of Medicine and Cardiovascular Research Institute and conformed to the guidelines of the American Heart Association. Surgical myocardial infarction was induced in 8-week-old male Sprague-Dawley rats (240 ± 10 g). Rats were anesthetized by intramuscular injection of ketamine hydrochloride (90 mg/kg) and xylazine hydrochloride (5 mg/kg). The anesthetized rats were ventilated with positive pressure (180 mL/min) by using a Harvard ventilator (Harvard Apparatus, Millis, MA). A 2-cm incision was made at the left lateral costal rib to expose the heart. The left anterior descending (LAD) artery was ligated for 1 h by using a 6-0 silk suture (Ethicon, Somerville, NJ), followed by reperfusion. Ischemia was confirmed by visual inspection of blanching in the myocardium distal to the occlusion site. In shamoperated rats, the same procedure was performed without LAD ligation and reperfusion. RAGE-siRNA/polyethyleneimine modified with deoxycholic acid (PEI-DA) nanocarriers were injected 1 h before surgical occlusion, and 3 intra-myocardial injections totaling 100 μL of RAGE-siRNA/PEI-DA nanocarriers (siRNA, 10 μM) solution in PBS were placed along the anterior and lateral aspects of the contracting wall bordering the infarct site. Rats were randomly divided into the following four groups: 1) control (sham operation, n = 10), 2) IR (IR only, n = 11), 3) IR+siRAGE (IR and RAGE-siRNA injection, n = 10), and 4) IR+scRNA (IR and scrambled siRNA injection, n = 9). Ambulatory Holter monitoring was performed using a telemetry system (Telemetry Research, Auckland, New Zealand).
Infarct Size and Fibrotic Area Analysis
To analyze the fibrotic area in cardiac tissues, heart sections were stained with conventional hematoxylin and eosin and Masson’s trichrome. The blue-colored fibrotic area was measured using MetaMorph software ver. 4.6 and was expressed as percent fibrosis (ratio of the fibrotic area to the total left ventricular area). To measure the infarct area, the hearts were sectioned transaxially and incubated in 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich, St Louis, MO) for 10 min at 37°C. The hearts were then fixed in 10% formalin overnight at 2-8°C and photographed using a digital camera. The viable myocardium was stained deep red, while infarct area was stained pale yellow. The infracted area was measured by planimetry by using the ImageJ software from the National Institutes of Health. The infarction rate was expressed as a percentage of infarction (total infarct size relative to left ventricular mass).
Optical Mapping
Rats (250-300 g) were anesthetized by intraperitoneal injection of ketamine (80 mg/kg) and xylazine (4 mg/kg). The chests were opened via median sternotomy, and the hearts were rapidly excised and immersed in cold Tyrode’s solution (composition: 125 mM NaCl, 4.5 mM KCl, 0.25 mM MgCl2, 24 mM NaHCO3, 1.8 mM NaH2PO4, 1.8 mM CaCl2, and 5.5 mM glucose). The ascending aorta was immediately cannulated and perfused with 37ºC Tyrode’s solution equilibrated with 95% O2 and 5% CO2 to maintain pH at 7.4. Coronary perfusion pressure was regulated between 80 and 95 mmHg. Two widely spaced bipolar electrodes were used for continuous pseudo-ECG monitoring.
For optical recording, the contractility of the heart was inhibited by 10-17 µM of blebbistatin [14]. The hearts were stained with di-4-ANEPPS (Invitrogen, Carlsbad, CA, USA) and excited with quasi-monochromatic light (520 ± 30 nm) from two green LED lamps. Emitted light was detected by a camera (MiCAM Ultima, Brainvision, Tokyo, Japan) with a 610-nm longpass filter. Data were gathered at 1-ms sampling intervals, acquired simultaneously from 100 × 100 pixels with spatial resolution of 0.3 × 0.3 mm2/pixel. The mapped area included parts of the right and left ventricular free walls.
Optical recordings were performed during steady-state and programmed stimulation. Programmed stimulation was performed with bipolar electrodes on the lateral side of the left ventricle. The pacing was initiated at a cycle length (CL) of 300 ms, by using stimuli of twice the pacing threshold, and reduced at decrements of 10 ms until 2:1 capture. Action potential duration (APD90) was measured at the base and apex of the left ventricle. APD dispersion is defined as the difference between maximum and minimum APDs. After the initial electrophysiological study, we attempted to induce ventricular tachycardia (VT) or ventricular fibrillation (VF) by using standard pacing techniques (burst pacing at CLs down to 70 ms). Sustained (>30 s) and nonsustained VT or VF episodes were documented. Optical mapping and VT induction studies were performed in 6 rats in each group.
Western Blotting
The rat hearts were homogenized before lysis. The protein concentration was determined using the Pierce BCA Protein Assay (Rockford, IL). The protein samples were separated by a SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membranes were blocked with 10% non-fat dry milk in 1× TBS containing 1% Tween 20 for 1 h. The membrane was incubated with indicated primary antibodies against β-actin (1:5000), RAGE (1:1000, Abcam, Cambridge, UK), tumor necrosis factor (TNF)-α (1:1000, Abcam), and anti-interleukin (IL)-6 (1:1000, Abcam) overnight at 4°C. After washing, primary antibodies were detected using horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG secondary antibodies. Detection was performed using an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, Piscataway, NJ).
Statistical Analysis
Data were expressed as the mean ± SEM. Student’s t-tests with Bonferroni correction were used to compare means of two numeric values. The Pearson's chi-square tests were used to compare two categorical variables. Using the paired t-test, we compared the means of maximum slope of the restitution curve between baseline and myocarditis. P values <0.05 were considered statistically significant.
Results
RAGE-siRNA Treatment Attenuated IR Injury
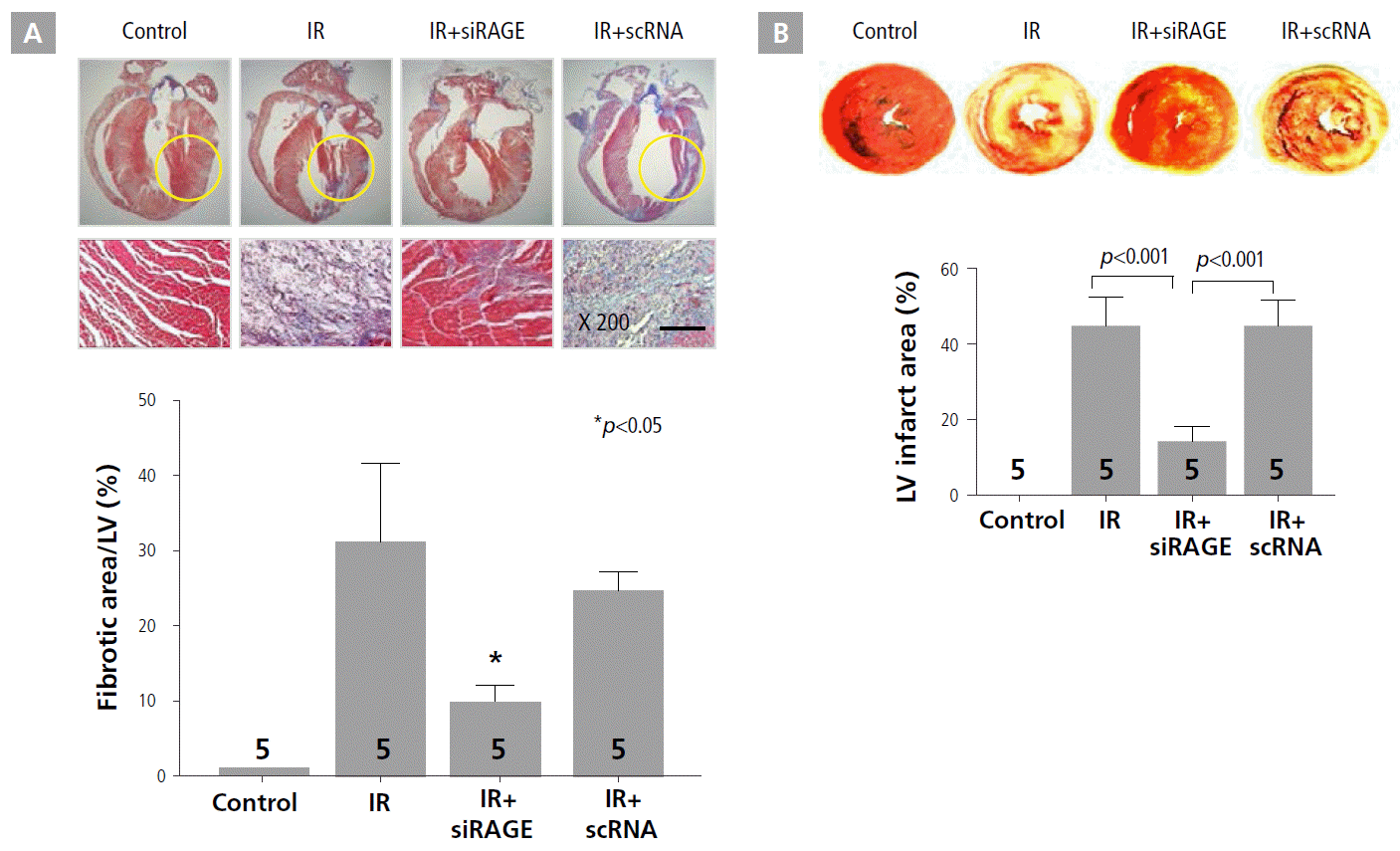
To evaluate cardioprotective effects of RAGE-siRNA nanocarriers against IR injury, the extent of myocardial fibrosis and infarct size were assessed by histology by using Masson’s trichrome staining and triphenyl tetrazolium chloride staining, respectively, at 7 days after treatment (Figure 1A and B). The quantification of the fibrotic area revealed a markedly lower degree of left ventricular fibrosis in the RAGE-siRNA nanocarrier treatment group compared to the IR and IR+scRNA groups, as shown in Figure 1A. Specifically, the fibrotic area was reduced in the IR+siRAGE group from 32% to 10% of the left ventricular wall. In addition, treatment with RAGE-siRNA nanocarriers caused significant attenuation of myocardial infarct expansion after LAD ligation in the rat MI model (Figure 1B). The IR+siRAGE group (14.2 ± 3.4%) showed a 3-fold reduction in degree of infarct expansion compared to IR (44.9 ± 7.5%) and IR+scRNA (44.9 ± 6.8%) groups.
RAGE-siRNA Treatment Improved Survival Rate and Decreased Inducible VT or VF in IR Rats
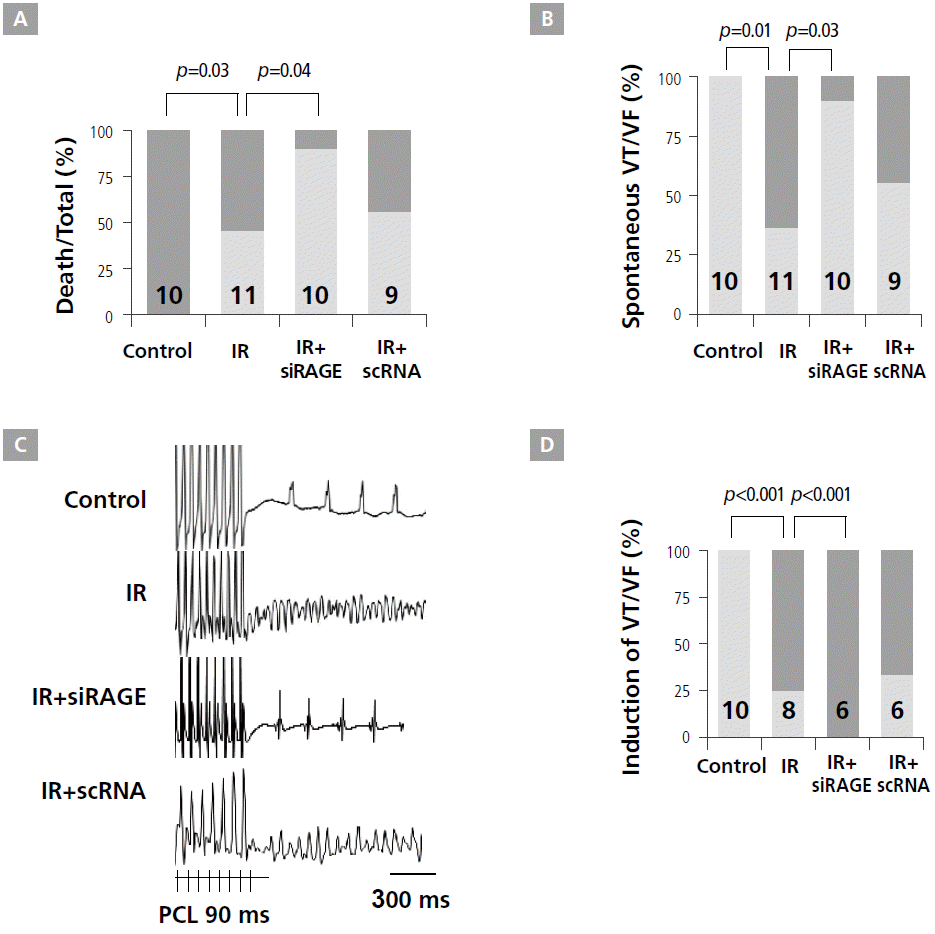
The results of short-term modality showed that the IR+siRAGE group had a significantly better heart survival rate than IR and IR+scRNA groups (Figure 2A). Specifically, in the IR group, 6 (50%) of 11 rats died at 46 ± 14 min after acute myocardial infarction. No rats died in control and IR+siRAGE groups. These results indicate that potential cardioprotective effects of the RAGE-siRNA nanocarriers can be attributed mainly to enhanced RAGE suppression, leading to a reduced innate immune response after acute myocardial infarction. While spontaneous VT or VF were not observed in control rats, they were observed in 6 (65%) rats in the IR group (p=0.01). However, only 1 (10%) rat in the IR+siRAGE group had VT, showing a significant reduction in VT episodes after siRAGE injection in IR rats (p=0.03, Figure 2B). Representative examples of induced VF are presented in Figure 2C. For the control (n=8), IR (n=8), IR+siRAGE (n=6), and IR+scRNA (n=6) groups, VT was induced in 0, 6 (75%), 0, and 4 (67%) rats, respectively. The IR group had higher inducible VT or VF than the control group (p=0.001). siRAGE prevented inducible VT or VF after MI (p=0.01). However, these protective effects were not observed in the IR+scRNA group (Figure 2D).
Effects of siRAGE Treatment on AP
The shape and time course of action potentials (APs) from the control, IR, IR+siRAGE, and IR+scRNA hearts are illustrated in Figure 3. In Langendorff perfused rat hearts, siRAGE improved the conduction time of LV. Figure 3A shows the activation map and action potential of 4 groups. Compared with controls, the total conduction times of ventricle were increased in the IR group from 11.0 ± 1.8 ms to 20.5 ± 2.4 ms (p<0.001). The IR+siRAGE group (16.8 ± 1.2 ms) showed a significantly decreased conduction time compared to the IR group (p=0.03).
These electric characteristics are shown for the normal zone, infarct border zone, and infarct zone in Figure 3. APD90 values were determined to be shorter in the IR group than in the control group (p<0.05), shorter in the IR group than in the IR+siRAGE group (p<0.05), and shorter in the IR+scRNA group than in the IR+siRAGE group (p<0.05) by using analyses of variance. Conduction velocity (CV) was slower in the IR and IR+scRNA groups than in the control group, and IR+siRAGE markedly increased CV compared with IR only and IR+scRNA (p<0.005).
Effect of siRAGE on Restitution Kinetics
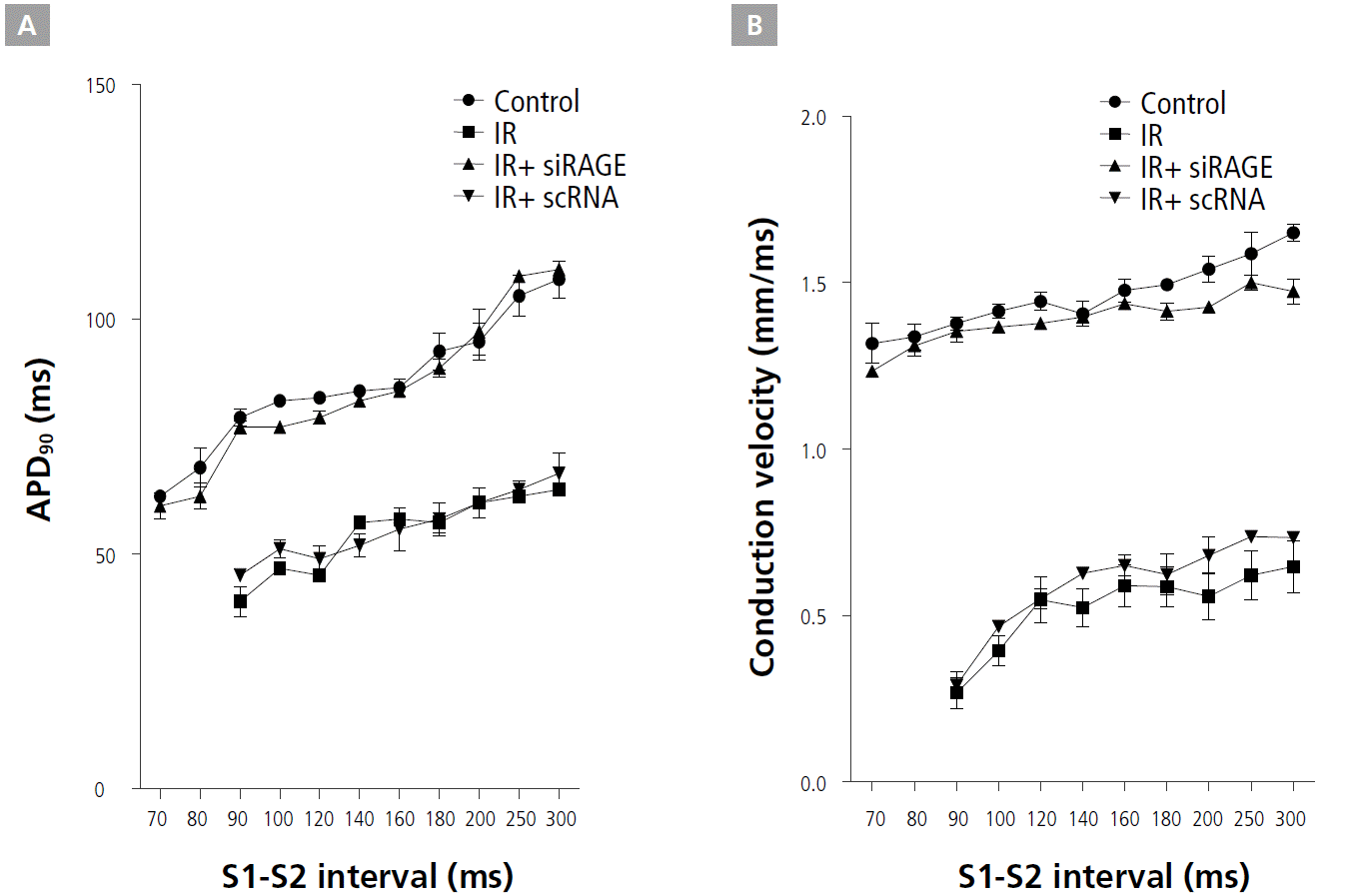
CV and APD restitution kinetics (RK) were measured from the infarct border zone of the control, IR, IR+siRAGE, and IR+scRNA groups. While IR flattened the APD RK curve, RAGE treatment reversed the slope of APD RK curves (Figure 4A). IR+siRAGE group showed decreased the maximum slope of APD restitution curve (0.34±0.09 vs. 1.14±0.26, p<0.001). siRAGE-treated IR hearts had longer APD90 RK curves compared with IR and IR+scRNA hearts, consistent with the APD90 shown in Figure 4A. Figure 4B shows a marked effect of RAGE on the CV RK compared with IR hearts, including a large increase in CV, particularly for short S1-S2 intervals and a less-steep RK curve. The slower CV of the premature pulse and of the first spontaneous reentrant beat in IR and IR+scRNA ventricles helped to sustain VT.
Increased VT Vulnerability in IR Model and Suppression by RAGE-siRNA Treatment
Figure 5 illustrates AP from an IR heart before and during a transient VT (Figure 5A) and during a sustained VF (Figure 5B). Activation maps during transient (a-g) VT (Figure 5A) exhibited a stable reentry pattern, with wavefronts emanating from a similar origin and propagating in a similar direction between beats. In contrast, during sustained VF (Figure 5Ba'-5Bg'), the origins of successive reentrant waves varied randomly and the arrhythmia was perpetuated by coexisting reentrant circuits maintained by the continuous annihilation and creation of daughter wavelets [15]. Voltage oscillations during VF were analyzed in a DF map (Figure 5C). The analysis showed that the normal zone (Figure 5C②) had higher dominant frequencies than the infarct border zone (Figure 5C①) (25.8 ± 3.0 vs. 19.0 ± 2.3 Hz, p=0.01, Figure 5D).
RAGE Suppression Reduced Inflammatory Responses in IR Rats
Since the upregulation of RAGE is tightly associated with the inflammatory response, cytokine expression patterns were further verified in a rat IR model treated with RAGE-siRNA nanocarriers (Figure 6). In general, at the site of inflammatory responses, interactions between RAGE and its various ligands caused the generation of proinflammatory cytokines such as IL-6 and TNF-α. Indeed, the levels of IL-6 and TNF-α were significantly increased above the normal range after acute IR. This enhanced production of the proinflammatory cytokines was dramatically reduced by RAGE-siRNA nanocarrier treatment, though negative-RAGE nanocarriers showed no anti-inflammatory effects in the rat IR model.
Discussion
The main findings of this study were that IR hearts showed elevated levels of RAGE and higher susceptibility to VF. The IR ventricle had a slower CV and higher level of fibrosis and inflammation. The RAGE-siRNA polyplex significantly reversed fibrosis and inflammation, increased ventricular CV, and suppressed VF.
Post-infarct Remodeling: Inflammation and Cardioprotection
Immediately after ischemic injury, various intra- and intercellular pathways contribute to healing of the myocardial wound in order to restore tissue integrity and function. Activation of the immune system after cardiac injury follows the pattern of immune activation after infection. Several inflammatory mediators are released and inflammatory cells are attracted to the site of injury. Thus, certain products of tissue injury, such as reactive oxygen species and intracellular proteins released from necrotic cells, initiate an inflammatory response, leading to the activation of pattern recognition receptors such as Toll-like receptors, the transcription nuclear factor kappa B, and complement factors.
RAGE is a key factor in myocardial IR injury [13], an indicator of chronic heart failure severity, and an independent marker of coronary artery disease and its severity in patients with congestive heart failure [16]. RAGE affects the generation of inducible nitric oxide synthase, nitrite and nitrate, and cGMP in the heart, particularly after IR injury. The pharmacological blockade of RAGE protects the isolated perfused heart from damage and dysfunction. In the mouse model, release of lactate dehydrogenase was attenuated in RAGE-null hearts. Furthermore, protection in RAGE-null mice hearts was associated with improved functional and metabolic recovery. However, the improvement in electrophysiological changes by cardioprotection was not fully elucidated. In this study, increased fibrosis, TNF-α, and IL-6 were observed in IR injury. The RAGE-siRNA polyplex significantly reversed fibrosis and inflammation.
Improvement of Myocardial Conduction by Cardioprotection Therapy with RAGE-siRNA
Changes in ionic currents caused by acute ischemia were shown to modify refractoriness and propagation velocity in cardiac tissue. The simulation showed that in the ischemic cell, K+ accumulation outside the cell had a major effect on cell excitability by depolarizing resting membrane potential, and reducing sodium channel availability. Increased post-repolarization refractoriness was caused by the slow recovery of the Na+ inactivation gates under conditions of resting depolarization. Moreover, CV was markedly slowed in the ischemic region. Slower conduction was caused by decreased INa resulting from slower recovery of the Na+ inactivation gates after resting depolarization of the cell membrane and then to decreased INa conductance caused by intracellular and extracellular acidosis. Ischemia-induced reduction of CV was remarkably improved after cardioprotection therapy with RAGE-siRNA.
Anti-arrhythmic Effects of Cardioprotection Therapy by RAGE-siRNA
APDs of ischemic cells did not exhibit rate adaptation characteristic, causing flattening of the APD restitution curve [17,18]. With the reduction in sodium channel availability and long recovery of excitability, the refractory period of ischemic tissue was significantly prolonged and could no longer be considered to be the same as APD. Therefore, global ischemia leads not only to a delayed but also to a non-uniform electrical restitution. The delay in electrical restitution may be causally related to the development of alternans of APD, whereas the dispersion of electrical restitution may produce electrical instability and set the stage for ischemia-related ventricular arrhythmias [17,18]. The slope of the CV restitution curve increased both in the normal and ischemic regions when pacing cycle length was short; moreover, refractory period dispersion increased with shortening of pacing cycle length [19]. Therefore, dynamic changes in CV and dispersion of the refractory period were suggested to be the fundamental mechanisms of arrhythmia in the regional ischemic myocardium. In this study, cardioprotection therapy by RAGE-siRNA reversed the changes in CV RK by IR injury.
Role of Heterogeneities in VF
Inflammatory cell infiltration and the myocardial structure destruction in the IR group were not observed in the IR+siRAGE group. The relative contribution of fixed versus dynamic heterogeneities in the mechanism of VF remains unclear [20,21]. Regional myocardial ischemia is a clinically relevant condition in which very large macroscopic heterogeneities occur naturally. Therefore, ischemia caused an increase in wavebreaks in the border zone and a decrease in wavebreaks in the ischemic zone, without a significant change in the nonischemic zone. This finding is consistent with the dominant-frequency distribution during ischemic VF: the average dominant frequency was significantly slower in the infarct zone than in the non-infarct zone, suggesting a large gradient in refractory periods across the border zone [22]. Cardioprotection therapy by RAGE-siRNA reduced tissue heterogeneity by reducing fibrosis and inflammation.
Conclusion
Our results revealed an association between RAGE and myocardial IR injury-associated cardiac dysfunction. The cardioprotection therapy with RAGE-siRNA polyplexes was effective in preventing fatal ventricular arrhythmia by reversing fibrosis and inflammation and increasing ventricular CV. We propose that RAGE-siRNA polyplexes combined with PE-DA can be used to exert cardioprotection in the heart, particularly after injury exacerbated by IR.