









Introduction
Myotonic dystrophy (dystrophia myotonica [DM]) is an autosomal-dominant, multi-systemic disease for which the clinical presentation includes myotonia, muscular dystrophy, cardiac involvement, posterior iridescent cataracts, and endocrine disorders.1 The incidence of myotonic dystrophy type 1 (DM1) has been estimated to be 1 case per 8000 births, and its worldwide prevalence ranges from 2.1 to 14.3 cases per 100 000 inhabitants.2 In patients with DM1, cardiac involvement (e.g., conduction abnormality and conduction blocks) is common; such involvement contributes significantly to DM1-related morbidity and mortality.3 However, only few cases of arrhythmic cardiac involvement with significant gastrointestinal abnormalities, especially in the esophagus, have been reported. Here, we describe a case of myotonic dystrophy and achalasia-like esophageal dilatation with atrial fibrillation (AF) treated with radiofrequency catheter ablation (RFCA). Myotonic dystrophy was confirmed by genetic testing.
Case
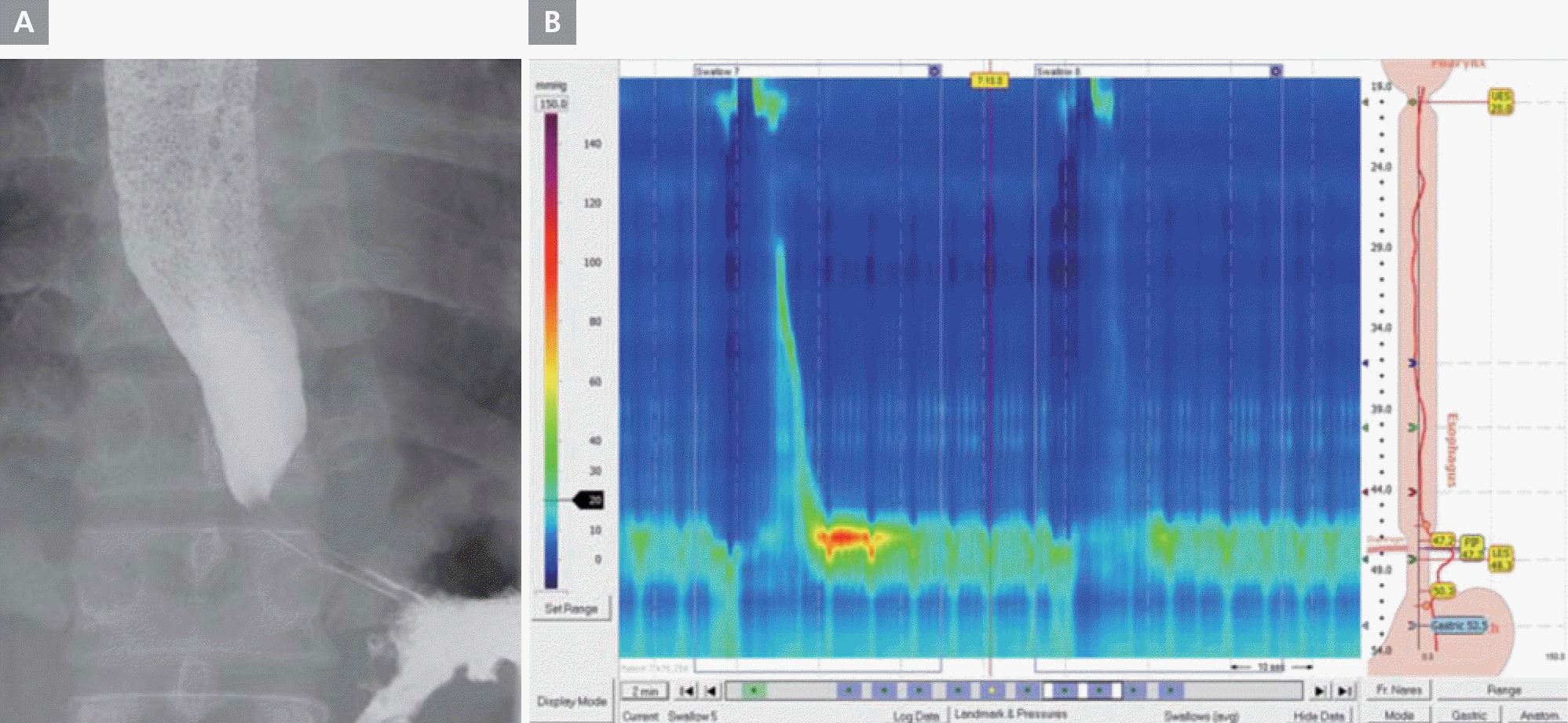
On March 7, 2009, a 46-year-old man presented with palpitation that had persisted for several years. He had undergone DC cardioversion for AF 8 months prior. He did not have a history of hypertension, diabetes mellitus, dyslipidemia, or ischemic heart disease; however, he had a stroke 4 years prior. His electrocardiogram (ECG) showed AF (Figure 1A), and his transthoracic echocardiogram showed normal left ventricular systolic function and no significant structural abnormalities, except for left atrial enlargement (4.1 cm) in the parasternal long-axis view. We decided to perform RFCA for the multi-drug-refractory recurrent AF. During the RFCA, baseline electrophysiological measurements indicated that the PR and HV intervals were prolonged by 273 and 89 msec, respectively. During the procedure, his gastrografin esophagogram showed extensive dilatation of the midbody of the esophagus, severe persistent narrowing at the esophago-gastric (EG) junction, and dye stagnation in the esophagus due to lack of peristaltic movement (Figure 2). We successfully performed 4 pulmonary vein antral ablations (Figure 3) and finished the procedure on June 23, 2010, with no safety issues. During the RFCA, the left pulmonary vein posterior antral ablation line was slightly close to the pulmonary vein trunk because of the markedly dilated midbody of the esophagus. After the RFCA, his routine barium esophagogram showed extensive dilatation of the esophageal lumen and narrowing of the EG junction, termed a “bird-beak appearance” (Figure 4A). We also performed high-resolution esophageal manometry, which revealed significantly decreased pressure in the upper esophageal sphincter and completely failed peristalsis. However, the pressure in the lower esophageal sphincter was normal (Figure 4B). Therefore, we confirmed the diagnosis of aperistalsis (absent contractility) even though the esophagogram had an achalasia-like shape. Although no peristaltic movement was present in the esophagus, the pressure in the lower sphincter did not increase. Therefore, conservative treatment was given. The patient's condition was tolerable, without any significant symptoms. Five years after the procedure, the patient had mild general muscle weakness and was further evaluated for systemic neuromuscular disease.
Figure 1.
A. ECG acquired on admission (June 17, 2010) showing atrial fibrillation. B. Follow up ECG acquired on January 20, 2017, showing sinus rhythm and PR prolongation.
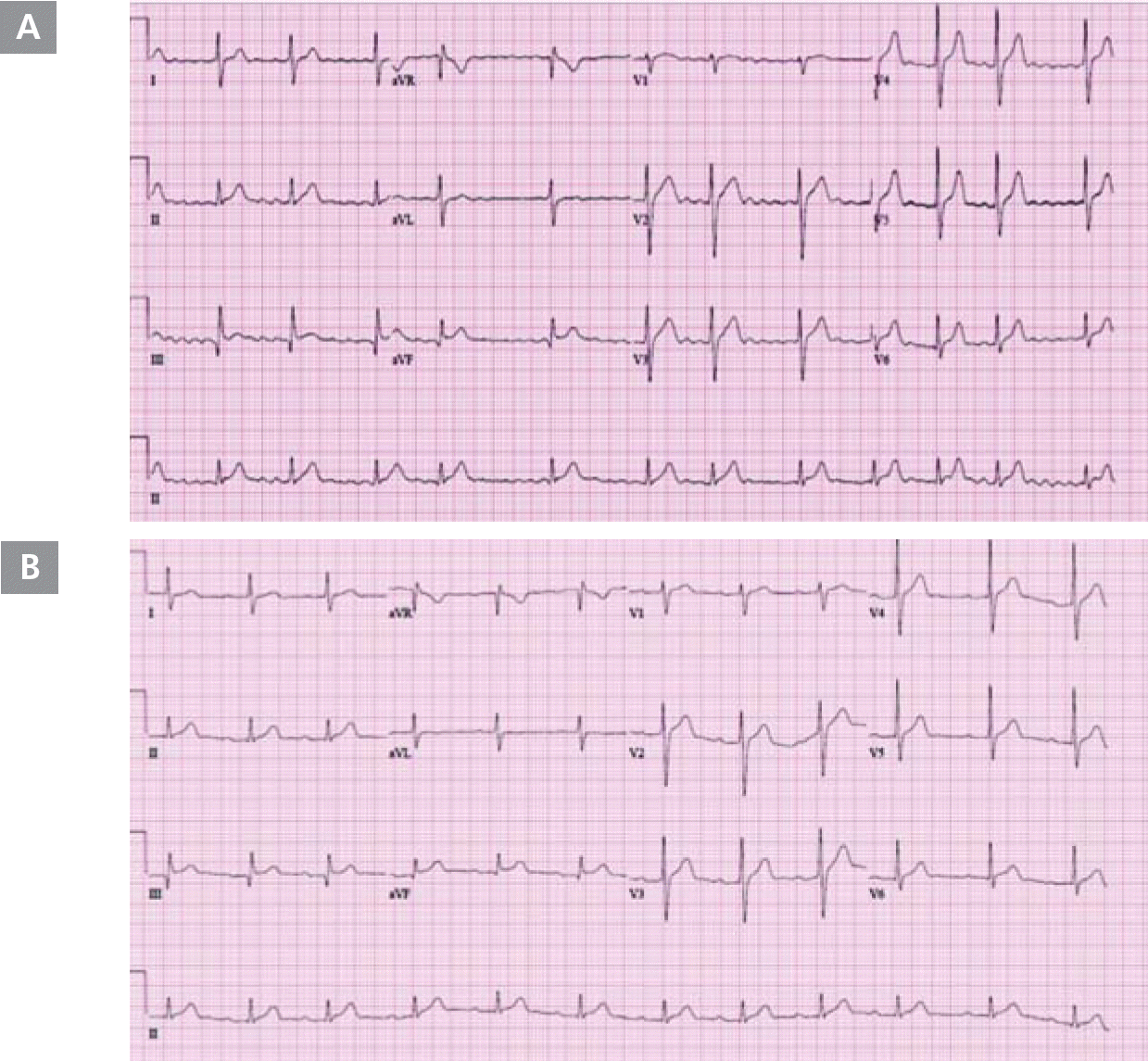
Figure 2.
Gastrografin esophagogram acquired during radiofrequency catheter ablation showing extensive dilatation of the midbody of the esophagus and severe narrowing at the esophago-gastric junction.
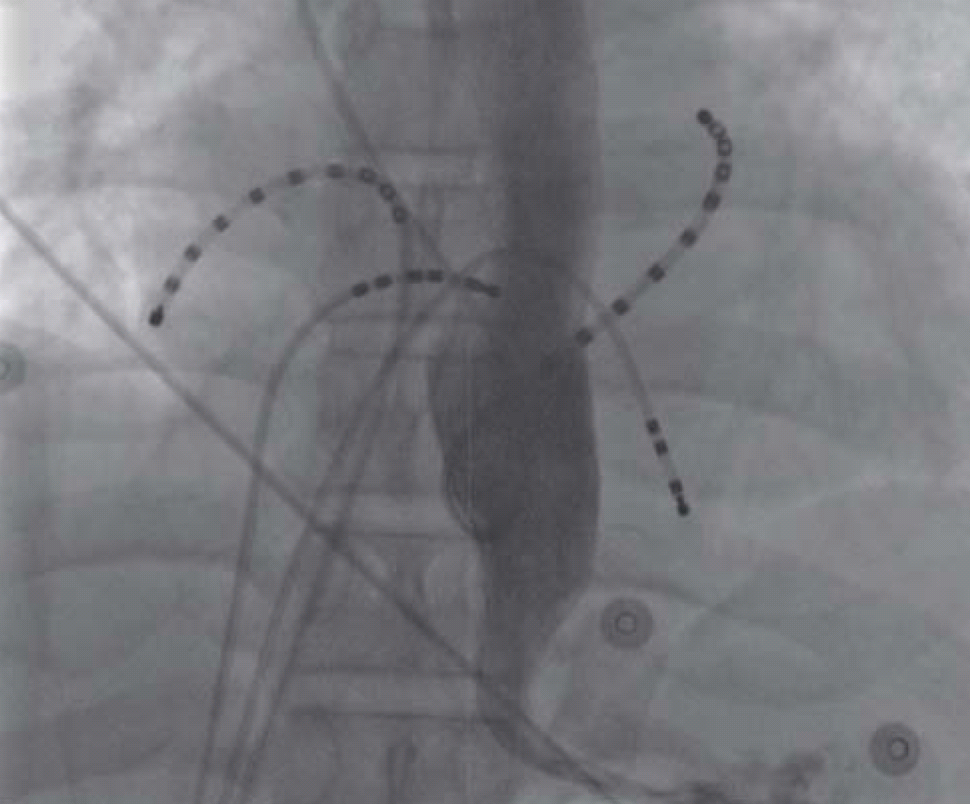
Figure 3.
Carto image after radiofrequency catheter ablation showing ablation lines of 4 pulmonary vein antrum.

Figure 4.
A. Barium esophagogram. Marked dilatation of the mid-esophagus with bird beak-like narrowing of the esophago-gastric junction. B. High resolution esophageal manometry showing aperistalsis. The patient showed significantly decreased pressure of the upper esophageal sphincter (mean basal pressure, 3.3 mmHg) and failed peristalsis with normal pressure of the lower esophageal sphincter (integrated relaxation pressure, 2.7 mmHg).
Genetic testing consisted of polymerase chain reaction (PCR) amplification of a region of the DM-protein kinase (DMPK) gene, including the cytosine-thymine-guanine (CTG) repeat region. Amplification was done using primers DM1-F (5′-FAM-GAAGGGTCCTTGTAGCCGGGAA-3′) and DM1-R (5′-GGAGGATGGAACACGGACGG-3′). To estimate the amplicon size, the GeneScan analysis program was used on an automated sequencer (ABI Prism 3130 Genetic Analyzer, Applied Biosystems). Southern blot analysis was performed to detect the larger amplicon corresponding to the larger allele of the CTG expansion, as previously described.2,4 The analysis identified repeats of approximately 500 (CTG) units in the 3′ untranslated region of the patient's DMPK gene, indicating classic DM1.
The patient has maintained sinus rhythm without taking any anti-arrhythmic agents thus far, 7 years after AF ablation (Figure 1B). Serial Holter monitoring showed no significant AF or ventricular tachycardia (VT). Although he currently experiences mild muscle weakness, he has no significant limitations in his daily life.
Discussion
DM is an autosomal-dominant inheritance disorder. Myotonia refers to impaired muscle relaxation after contraction and is caused by repetitive myocyte depolarization. Patients complain of muscle stiffness such as difficulty in releasing a handgrip after a handshake.
DM is of two main types; type 1 (DM1) is caused by mutations in the DMPK gene, whereas type 2 (DM2) is caused by mutations in the cellular nucleic acid-binding protein (CNBP) gene. Diagnosis is confirmed by genetic testing.1 Conduction system abnormalities are commonly observed in DM1. Fibrosis and fatty infiltration are observed together in the conduction system and might be part of the underlying mechanism of the development of conduction system defects.5 Cardiac involvement is mainly because of selective degeneration of the infranodal conduction system.6 While the His-Purkinje system is most frequently involved, any part of the conduction system can be affected.7 In patients with DM1, atrial flutter and atrial fibrillation are common supraventricular arrhythmias and observed in approximately 25% of all patients.7 Ventricular arrhythmias are also frequent. VT and ventricular fibrillation have been continuously reported.8
Our patient had esophageal motility disorder in addition to aperistalsis. While he complained of mild impairment of swallowing, his symptoms were not severe; therefore, we did not perform any surgical treatment. Steinert suggested an esophageal involvement in DM and reported that swallowing disturbance occurs in one-third of all such patients.9
Only one case of DM with cardiac involvement has been reported in Korea, which involved a patient with myotonic dystrophy with prolonged atrial flutter.10 To the best of our knowledge, this is the first report of RFCA in a patient with persistent AF with DM and achalasia-like esophageal dilatation due to aperistalsis.



