|
|
International Journal of Arrhythmia 2014;15(1): 6-19.
|
 |
| ORIGINAL ARTICLE |
Losartan attenuates atrial structural
and electrical remodeling in rat ischemic
heart failure model: Implications of
endothelin for atrial fibrillation |

|
Kyoung-Im Cho, MD, Soo-Jeong Lee, MD, Tae-Joon Cha, MD, PhD, Sang-Ho Koo, MS, Jung-Ho Heo, MD, PhD, Hyun-Su Kim, MD, and Jae-Woo Lee, MD, PhD
Division of Cardiology, Department of Internal Medicine, Kosin University College of Medicine, Busan, Korea
|
|
|
|

Introduction
Atrial fibrillation (AF) is the most common form
of sustained arrhythmia. It is associated with cardiac dysfunction and thrombus formation, two major
complications resulting in an increased risk of morbidity
and mortality from congestive heart failure
(CHF) and stroke.1,2 AF is commonly associated with
CHF, because CHF creates the conditions necessary
for AF development via structural remodeling and
neurohormonal remodeling.3 Therefore, elucidating
the exact role that CHF plays in the development AF
is of paramount importance.
There are two mechanisms of arrhythmia development
in CHF. One mechanism is reentry, which is
associated with increased tissue fibrosis4 due to activation
of the renin-angiotensin system (RAS).5 In
a nonclinical study, it was demonstrated that angiotensin
II receptor blockers (ARB) prevent AF by
reducing atrial structural remodeling.6 The other
important mechanism of arrhythmia development
in CHF is triggered activity.7 By performing biatrial
mapping experiments in dogs with CHF, Fenelon
et al. demonstrated that the majority of AF episodes
involved a focal mechanism. Another anti-AF mechanism
of ARBs may be the prevention of focal electrical
discharge.8
Multiple neurohormonal factors, including endothelin-1 (ET-1), are altered in patients with CHF.
ET-1 concentration is increased in cardiac tissues
during pathological conditions such as CHF9 and
myocardial infarction (MI).10 ET-1 can provide short-term
inotropic support for failing hearts, however,
this benefit comes with the potential burden of arrhythmogenesis
and remodeling.11 The arrhythmogenic
effect of ET-1 is mediated by activation of
inositol 1,4,5-triphosphate.12 ET receptor antagonists
have shown promising results in animal models of
CHF,13,14 but have failed to improve morbidity and
mortality in clinical trials in humans.15 Recent reports
have suggested that angiotensin II is a powerful
stimulator of ET synthesis and release in vascular smooth muscle and endothelial cells.16,17 Therefore, in
this study, we investigated the role of the ET system
in the development of AF in a model of CHF. We
also evaluated the potential AF-suppressing effect of
ARBs to determine whether blocking ET-1 synthesis
is of significance.
Materials and Methods
Animal preparation
All animal experiments were performed in accordance
with Korean Council on Animal Care guidelines,
and under the authority of the Animal Research Ethics
Committee of the Kosin University School of Medicine.
Male Sprague-Dawley (SD) rats were purchased from
Daehan Biolink Inc. (Korea). Rats were 7-8 weeks old
and weighed between 250 and 300 g. Rats were fed a
normal sodium diet and offered tap water ad libitum.
MI was induced by ligation of the left anterior descending
coronary artery (LAD) as described in detail
previously.18 Rats were allowed to recover for 2 days
and then randomized into 3 groups: sham (n=20), MI
(n=19), and MI + losartan (n=20). Losartan (10 mg/kg/day) was added to the drinking water and administered
for a period of 10 weeks. Water consumption
and body weight were carefully monitored. Sham operated
rats served as the control group.
MI procedure
Rats were anesthetized by i.p. administration of 50
mg/kg ketamine and 10 mg/kg xylazine. Rats were
ventilated with room air at 60 strokes per min and
a stroke volume of 1 mL/100 g body weight (Harvard,
Rodent Ventilator, Model 683). After opening
the chest by mid-sternotomy, the LAD was ligated with a 5.0 polypropylene suture (Ethicon, Belgium)
approximately 2 to 3 mm from its origin. Control rats
were subjected to a sham operation using a similar
procedure, but without coronary ligation. Perioperative
mortality was around 40% in rats subjected to
coronary artery ligation.
Echocardiography
At the end of the study, rats were lightly anesthetized
by i.p. administration of 25 mg/kg ketamine
with 5 mg/kg xylazine (n=10/group). Transthoracic
echocardiography was performed using an Acuson
Sequoia C512 12-MHz phased-array probe (Mountain
View, CA, USA). Wall thickness and left ventricular
(LV) diameters during diastole and systole were
measured from M-mode recordings according to the
leading-edge method. Two-dimensional, short-axis
images at the mid-papillary muscle level were obtained
for measurement of LV end-diastolic dimension
(LVEDD), LV end-systolic dimension (LVESD),
fractional shortening (FS), and wall thickness. Ejection
fraction (EF) was calculated as follows: EF =
(LVEDD2- LVESD2)/LVEDD2×100. Left atrial (LA)
and aortic diameters were measured from M-mode
recordings in a modified, parasternal, long-axis view.
Electrophysiological study
On the open-chest study days, rats were anesthetized
with ketamine (90 mg/kg) and xylazine (13 mg/
kg) and ventilated mechanically. A sternotomy was
performed, and bipolar electrodes were hooked into
the right-atrial appendage for recording and stimulation.
AF was induced by burst pacing (10 Hz, 4 times
threshold, 30 seconds). Mean AF duration was estimated
by the average of 10 inductions if AF duration
was ≤20 min, and from 5 inductions if AF duration lasted between 20 and 30 min. AF lasting longer than
30 minutes was considered persistent, and in this
case, mean AF duration was recorded as 30 minutes.
Quantification of fibrosis
After completion of the electrophysiological study,
hearts were rapidly excised and weighed in order to
calculate heart:body weight ratio. Hearts were immersed
in a 10% buffered formalin solution and then
embedded in paraffin. Sections were stained with
Masson’s trichrome staining to characterize the collagen
fibers. Fibrous tissue content of the LA was analyzed
with Image-Pro Plus 5.1 (Media Cybernetics,
GA, USA), and quantified as percent surface area,
excluding blood vessel-containing regions.
RNA purification
Total RNA was isolated from sham, MI, and MI +
losartan LA tissues (10-20 mg) with Reboex reagent
(Genall, Korea), followed by chloroform extraction
and isopropanol precipitation. Genomic DNA was
eliminated by incubation in rDNase I (2 U/μL, 37℃,
Ambion, USA) for 30 minutes. RNA was quantified
by spectrophotometric absorbency at a wavelength
of 260 nm; purity was confirmed by calculating the
A260/A280 ratio.
Real-time RT-PCR
Gene-specific primers for real-time reverse transcription
(RT)-polymerase chain reaction (PCR) were
designed based on published cDNA sequences for rat
ET-1 (Table 1). First-strand cDNA synthesized by RT
of rat atrial mRNA samples was used as a template
for subsequent RT-PCR experiments. Two-step RT
PCR was conducted with LightCycler® Real-Time RT-PCR system (Roche Applied Science). Primers
used for the detection of ET-1 and GAPDH are
shown in Table 1. RT PCR was run in the presence of
a double-stranded DNA binding dye (SYBR). GAPDH
was used as an internal standard, and all ET-1 results
were normalized to GAPDH data obtained from
the same samples at the same time. Total RNA was
run in duplicate for each experiment.
Western blot
LA tissue (~25 mg) was homogenized in a modified
tonic sucrose solution (0.3 mol/L sucrose, 10 mmol/L
imidazole, 10 mmol/L sodium metabisulfite, 1 mmol/L
DTT, 0.3 mmol/L PMSF), and centrifuged at 1300 xg
for 15 min at 4°C. Extracted protein was quantified
by Bradford assay. Aliquots containing 50 μg ETaR
and ETbR were separated with 13%-polyacrylamide
gel-SDS electrophoresis (90 min), then transferred
to PVDF membrane (Pierce, USA). Immunoblotting
was performed with ETa and ETb polyclonal antibodies
(Alamone, USA), at a 1:500 dilution in 5% milk/
TBST. GAPDH (1:5000, Santa Cruz, USA) was used
as control for protein loading. Immunoreactive bands
were visualized by SuperSignal West Pico Chemiluminescent
Substrate (Pierce, USA) and exposed to X-ray
film. Densitometric analysis of western blots was
performed, and band intensities are expressed relative
to GAPDH intensity from the same sample.
Immunohistochemistry for ET-1, ETa, and ETb
Additional hearts were used for the immunohistochemical
visualization of ET-1, ETaR, and ETbR.
Hearts were dissected, fixed for 3 h in 4% phosphatebuffered
paraformaldehyde solution (pH 7.4), and
processed for embedding in paraffin. Sections (6-μm
thickness) were cut, mounted on silane-coated slides,
and heated to 60°C for 35 min. Slides were then deparaffinized
in xylene, and rehydrated in graded
immunoglobulin-free bovine serum albumin (BSA,
Sigma, USA) in PBS for 20 min at room temperature.
Slides were subsequently incubated for 2 h at room
temperature with rabbit anti-ET-1 (1:50, Peninsula
laboratories), rabbit anti-rat-ETa (1:50, Alamone,
USA), and rabbit anti-rat-ETb (1:50, Alamone,
USA) polyclonal antibodies diluted in 3% BSA. Sections
were washed once in 0.1% Triton X-100, and
twice in PBS. Antibody binding was detected using
anti-rabbit and anti-mouse UltraVision LP Detection
System (Lab Vision Corporation, Suffolk, UK),
according to the manufacturer’s instructions.
Drugs
Lorsartan (a gift from Merck Pharmaceuticals,
Seoul, Korea) was dissolved at strength in vehicle.
Statistical Analysis
Data were expressed as mean ± SEM unless otherwise
indicated. Kruskal-Wallis tests were used for
statistical comparisons. If Kruskal-Wallis tests were
significant, group-to-group comparisons were performed
using the Mann-Whitney test. Differences
were considered statistically significant when p<0.05.
Analyses were performed with SPSS 12.0 (SPSS Inc.,
Chicago, IL, USA).
RESULTS
Echocardiographic indices
Echocardiographic parameter changes are presented
in Figure 1 and Table 2. LV EF was significantly
lower in the MI group compared with the sham
group (32.3 ± 4.3 vs. 84.8 ± 2.7%, p<0.01). However,
LV EF was significantly higher in the MI + losartan
group compared with the MI group (47.8 ± 4.4 vs. 32.3 ± 4.3%, p<0.05).

AF induction study
AF inducibility was significantly higher in the MI
group compared with sham controls (22.6 ± 5.3 vs.
5.5 ± 2.2%, p=0.001). Treatment with losartan significantly
lowered AF inducibility compared with the
MI group (13.7 ± 5.3% vs MI, p=0.038). Mean AF duration significantly increased in MI rats compared
with sham controls (293.2 ± 153.8 vs. 9.8 ± 7.4 seconds,
p=0.001), and was moderately reduced after
losartan treatment (182.1 ± 123.7 seconds, p=0.022
vs. MI) (Figure 2 and 3).


Preventive effects of losartan on LA fibrosis
Interstitial fibrosis in the LA was significantly increased in the MI group compared with the sham
group (n=5/group, 2.37 ± 0.30 vs. 0.24 ± 0.03%,
p=0.008). After treatment with losartan, the level of
LA fibrosis was significantly decreased compared with
the MI group (0.99 ± 0.12% in MI + losartan group,
p=0.008 vs. MI group) (Figure 4 and 5).

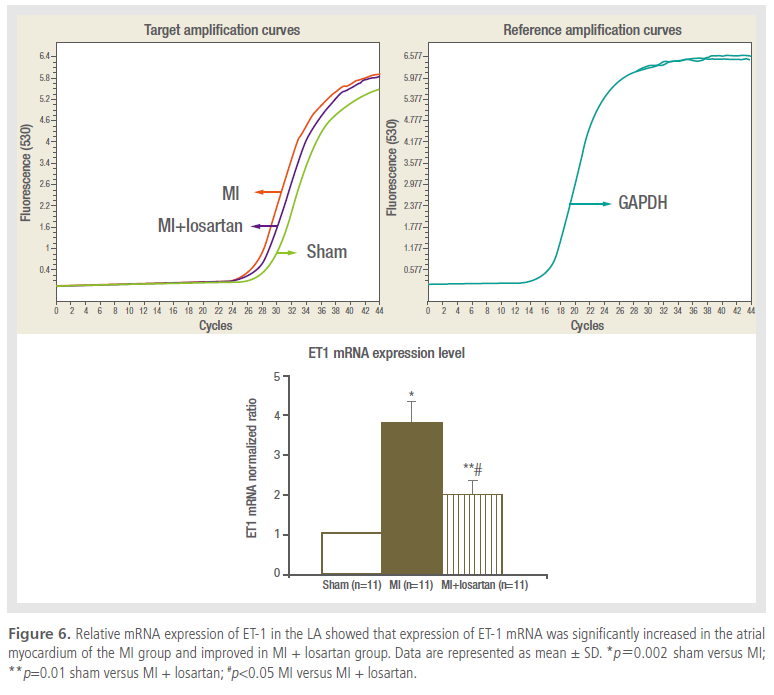
Change in transcription level of ET-1
The expression of ET-1 mRNA in LA tissue of rats
from the MI group was ~3.5 times upregulated compared
with the sham group (p=0.002). This change recovered slightly in rats that received losartan treatment
(p=0.023), as shown in Figure 6.
Expression of ETa receptor and ETb receptor proteins
Protein level of ETaR in LA tissue was significantly
increased in the MI group as compared to the sham
group (p=0.002), and it was almost abolished in the
losartan-treated group (p=0.026). In contrast, protein
expression of ETbR in LA tissue significantly decreased in the MI group, but recovered to 80% of the
sham group level following treatment with losartan
(p=0.004) (Figure 7).

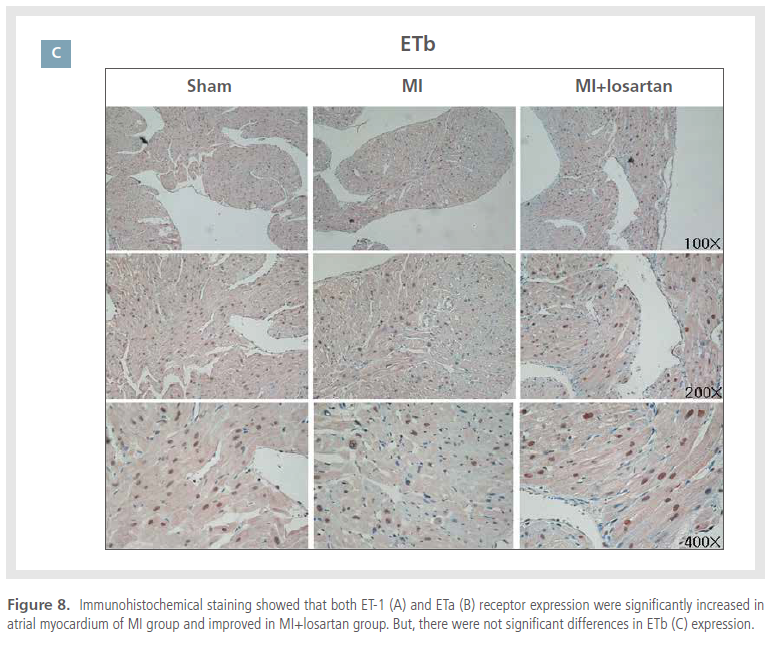
Immunohistochemistry results
Immunoreactive ET-1 and ETaR were rarely detected
in cardiomyocytes obtained from sham controls.
However, expression of ET-1 and ETaR were
increased in the MI group, which was prevented with
losartan treatment. Immunohistochemical staining
for ETbR showed some degree of ETbR activity in the
sham group, but myocardial ETbR activity was lower in the MI group (Figure 8). In general, immunohistochemical
results correlated with those of western
blot analysis.

DISCUSSION
This study demonstrated that expression of ET-1
and ETaR significantly increased and expression
of ETbR significantly decreased in the LA tissue of
MI-induced CHF rats. These observations were accompanied
by increased AF inducibility and increased
mean AF duration in the same model; thus implicating the ET system in the promotion of AF. After
treatment with losartan, these changes observed in
the ET system and AF development were reversed.
Together, these findings suggest a possible mechanism
to explain the anti-AF effects of ARBs in an
animal model of CHF.
The endothelin system in CHF
The ET system, a vascular regulatory system, has
received considerable attention in recent years.9-17 Of
the 4 active ETs, ET-1 is the predominant isoform
in the cardiovascular system and exerts its effects
through activation of 2 distinct G-protein-coupled receptors, ETaR and ETbR.19,20 ETaRs are found in
the medial smooth muscle layers of blood vessels, and
the myocardium of the atria and ventricles. When
stimulated, ETaRs induce vasoconstriction and cellular
proliferation by increasing intracellular calcium.21
ETbRs are localized in endothelial cells and, to some
extent, smooth muscle cells and macrophages.22 Activation
of ETbRs stimulates the release of nitric oxide
and prostacyclin, and prevents apoptosis.23 ETbRs
are down-regulated in CHF and contribute to neurohormonal
activation, hemodynamic deterioration,
and cardiovascular remodeling.24 Elevated plasma
ET-1 and big ET-1 levels have been reported after
spontaneous and triggered non-sustained supraventricular
and ventricular tachycardias. Dezsi et al. reported
significantly decreased plasma ET-1 levels
after catheter ablation of tachyarrhythmias.25 In the
present study, we demonstrated that ET-1 and ETaR
density increased in LA tissues of rats with MI-induced
CHF, which might be linked with the induction
of AF and increased duration of AF.
Relationship between angiotensin II, AF, and ET-1
Increased atrial interstitial fibrosis has been demonstrated
in patients with AF.26,27 Furthermore, elevated
quantities of interstitial fibrosis have been
shown to increase AF vulnerability in animal models
of CHF4,26-29 and in a transgenic model for selective
atrial fibrosis.30 ARBs prevent the promotion of AF by reducing atrial fibrosis,5 and the LIFE study
has shown that ARBs prevent subsequent AF development
in patients with hypertension.31 Based on
these studies, the preventive effects of ARBs on AF
are thought to be due to an attenuation of structural
remodeling that can lead to atrial interstitial fibrosis.
CHF is known to facilitate the release of angiotensin
II, which activates the secretion of ET-1 from
various tissues. We demonstrated increased mRNA
and immunoreactivity of ET-1 in MI-induced CHF
rats (Figure 6 and 8), and conclude that this could
be due to activation of angiotensin II and/or toxicity
of CHF. Along with increased ETaR expression,
ET-1 binding to ETaR could cause vasoconstriction
of heart vessels, proliferation of cardiomyocytes and/
or non-cardiomyocytes in atrial tissue, and activate
intracellular inositol 1,4,5-trisphosphate receptors.
These receptors trigger spontaneous Ca2+ increase,12
resulting in increased AF inducibility and atrial fibrosis.
Losartan treatment in MI-induced CHF rats
partially reversed the expression of ET-1 and EtaR,
and lessened subsequent AF development and LA fibrosis.
Therefore, it is postulated that the effects of
ET-1 on MI-induced CHF are most likely mediated
through angiotensin II. The role of ETbR in the heart
is less well known than that of EtaR. However, decreased
ETbR was consistent with decreased tissue
nitric oxide level in MI-induced CHF rats, which was
subsequently reversed with losartan treatment.
Limitations of study
In the present study, we did not directly measure
angiotensin II levels in LA tissue; however, increased
angiotensin II levels have already been documented
in CHF. Furthermore, the direct relationship between
elevation of ET-1 and arrhythmogenesis was not
evaluated using an ET receptor blocker. Bosentan, an ET receptor blocker, failed to demonstrate efficacy
in a CHF clinical trial, and is currently only used for
the treatment of primary pulmonary hypertension.
Since bosentan is not clinically relevant to CHF, we
did not evaluate the antiarrhythmic effects of such
direct ET-1 blockade in our CHF model. Moreover, ET 1-induced arrhythmogenesis has been reported
several times in the literature,32-34 therefore we did
not investigate ET-1-induced arrhythmogensis in
our study.
Acknowledgements
We thank Merck Pharmaceuticals for providing
losartan.
Disclosure Statement
The authors state no conflict of interest.
References
- Hart RG, Halperin JL. Atrial fibrillation and stroke: concepts and
controversies
Stroke.
2001;32:803-808.
- Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, Singer
DE. Prevalence of diagnosed atrial fibrillation in adults: national
implications for rhythm management and stroke prevention: the
AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA)
Study
JAMA.
2001;285:2370-2375.
- Hsu LF, Jais P, Sanders P, Garrigue S, Hocini M, Sacher F, Takahashi Y,
Rotter M, Pasquie JL, Scavee C, Bordachar P, Clementy J, Haissaguerre
M. Catheter ablation for atrial fibrillation in congestive heart failure.
N Engl J Med.
2004;351:2373-2383.
- Cha TJ, Ehrlich JR, Zhang L, Shi YF, Tardif JC, Leung TK, & Nattel S.
Dissociation between ionic remodeling and ability to sustain
atrial fibrillation during recovery from experimental congestive
heart failure.
Circulation.
2004;109; 412-418.
- Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S.
Effects of angiotensin-converting enzyme inhibition on the
development of the atrial fibrillation substrate in dogs with
ventricular tachypacing-induced congestive heart failure.
Circulation.
2001;104:2608-2614.
- Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. and structural remodeling in atrial fibrillation.
Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation.
J Am Coll Cardiol.
2003;41:2197-2204.
- Stmbler BS, Fenelon G, Shepard RK, Clemo HF, Guiraudon CM.
Characterization of sustained atrial tachycardia in dogs with
rapid ventricular pacing-induced heart failure
J Cardiovasc Electrophysiol.
2003;14:499-507.
- Fenelon G, Shepard RK, Stambler BS. Focal origin of atrial tachycardia
in dogs with rapid ventricular pacing-induced heart failure.
J Cardiovasc Electrophysiol.
2003;14:1093-1102.
- Duru F, Barton M, Luscher TF, Candinas R. Endothelin and cardiac
arrhythmias: do endothelin antagonists have a therapeutic potential
as antiarrhythmic drugs?
Cardiovasc Res.
2001;49:272-280.
- Russell FD, Molenaar P. The human heart endothelin system:
ET-1 synthesis, storage, release and effect.
Trends Pharmacol Sci.
2000;21:353-359.
- Zolk O, Munzel F, Eschenhagen T. Effects of chronic endothelin-1
stimulation on cardiac myocyte contractile function.
Am J Physiol Heart Circ Physiol.
2004;286:H1248-H1257.
- Proven A, Roderick HL, Conway SJ, Berridge MJ, Horton JK, Capper
SJ, Bootman MD. Inositol 1,4,5-trisphosphate supports the
arrhythmogenic action of endothelin-1 on ventricular cardiac
myocytes.
J Cell Sci.
2006;119:3363-3375.
- Mulder P, Richard V, Derumeaux G, Hogie M, Henry JP, Lallemand
F, Compagnon P, Mace B, Comoy E, Letac B, Thuillez C. Role of
endogenous endothelin in chronic heart failure: effect of long-term
treatment with an endothelin antagonist on survival, hemodynamics,
and cardiac remodeling.
Circulation.
1997;96:1976-1982.
- Sakai S, Miyauchi T, Kobayashi M, Yamaguchi I, Goto K, Sugishita Y.
Inhibition of myocardial endothelin pathway improves long-term
survival in heart failure.
Nature.
1996;384:353-355.
- Kelland NF, Webb DJ. Clinical trials of endothelin antagonists
in heart failure: a question of dose?
Exp Biol Med (Maywood).
2006;231:696-699.
- Sung CP, Arleth AJ, Storer BL, Ohlstein EH. Angiotensin type 1
receptors mediate smooth muscle proliferation and endothelin
biosynthesis in rat vascular smooth muscle
Pharmacol Exp Ther.
1994;271:429-437.
- Imai T, Hirata Y, Emori T, Yanagisawa M, Masaki T, Marumo F.
Induction of endothelin-1 gene by angiotensin and vasopressin in
endothelial cells.
Hypertension.
1992;19:753-757.
- Boixel C, Gonzalez W, Louedec L. Hatem SN. Mechanism of the down
regulation of the L-type calcium current in atrial myocytes of rat
in heart failure.
Circ Res.
2001;89:607-613.
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M,
Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor
peptide produced by vascular endothelial cells.
Nature.
1988;332:411-415.
- Levin ER. Endothelins.
N Engl J Med.
1995;333:356-363.
- Hosoda K, Nakao K, Hiroshi A, Suga S, Ogawa Y, Mukoyama M,
Shirakami G, Saito Y, Nakanishi S, Imura H. Cloning and expression of human endothelin-1 receptor cDNA.
FEBS Lett.
1991;287:23-26.
- Ogawa Y, Nakao K, Arai H, Nakagawa O, Hosoda K, Suga S,
Nakanishi S, Imura H. Molecular cloning of a non-isopeptide-selective
human endothelin receptor.
Biochem Biophys Res Commun.
1991;178:248-255.
- Niwa Y, Nagata N, Oka M, Toyoshima T, Akiyoshi H, Wada T,
Nakaya Y. Production of nitric oxide from endothelial cells by
31-amino-acid-length endothelin-1, a novel vasoconstrictive
product by human chymase.
Life Sci.
2000;67:1103-1109.
- Fukuchi M, Giaid A; Expression of endothelin-1 and endothelin
-converting enzyme-1 mRNAs and proteins in failing human
hearts.
J Cardiovasc Pharmacol.
31 1998:S421-S423.
- Dezsi CA, Szucs A, Szucs G, Roka A, Kiss O, Becker D, Merkely B.
Short-term effect of rate control on plasma endothelin levels of
patients with tachyarrhythmias.
Exp Biol Med.
2006;231:852-856.
- Frustaci A, Chimenti C, Bellocci F, Morgante E, Russ MA,
Maseri A. Histological substrate of atrial biopsies in patients
with lone atrial fibrillation.
Circulation.
1997;96:1180-1184.
- Kostin S, Klein G, Szalay Z, Hein S, Bauer EP, Schaper J. Structural
correlate of atrial fibrillation in human patients.
Cardiovasc Res.
2002;54:361-379.
- Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by
heart failure in dogs: atrial remodeling of a different sort.
Circulation.
1999;100:87-95.
- Lee KW, Everett TH, Rahmutula D, Guerra JM, Wilson E, Ding C,
Olgin JE. Pirfenidone prevents the development of a vulnerable
substrate for atrial fibrillation in a canine model of heart failure.
Circulation.
2006;114:1703-1712.
- Verheule S, Sato T, Everett T, Engle SK, Otten D, Rubart-von der
LM, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased
vulnerability to atrial fibrillation in transgenic mice with selective
atrial fibrosis caused by overexpression of TGF-beta1.
Circ Res.
2004;94:1458-1465.
- Wachtell K, Lehto M, Gerdts E, Olsen MH, Hornestam B, Dahlof B,
Ibsen H, Julius S, Kjeldsen SE, Lindholm LH, Nieminen MS, Devereux
RB. Angiotensin II receptor blockade reduces new-onset atrial
fibrillation and subsequent stroke compared to atenolol: the Losartan
Intervention For End Point Reduction in Hypertension (LIFE) study.
J Am Coll Cardiol.
2005;45:712-719.
- Yorikane R, Koike H. The arrhythmogenic action of endothelin in
rats.
Jpn J Pharmacol.
1990;53:259-263.
- Zhao X, Fu WJ, Yuan WJ, Hou GX, Chen JG, Xia JH, Zhu HN. In
fluence of endothelin-1 on ventricular fibrillation threshold in
acute myocardial ischemic rats.
Zhongguo Yao Li Xue Bao.
1994;15:363-366.
- Ding Y, Zou R, Judd RL, Zhong J. Endothelin-1 receptor blockade
prevented the electrophysiological dysfunction in cardiac myocytes
of streptozotocin-induced diabetic rats.
Endocrine.
2006;30:121-127.
|
|
|
|











